Früh- und Neugeborenenchirurgie
Ösophagusatresie
Ösophagusatresie
Symptome:
Ösophagusatresie ist eine angeborene Fehlbildung, bei der eine Unterbrechung der Speiseröhre im Vordergrund steht: Entweder hat die Speiseröhre keine Verbindung zum Magen und mündet in die Luftröhre oder sie hat eine so starke Verengung (Stenose), dass keine Nahrung passieren kann. Die verschieden Formen der Ösophagusatresie werden nach VOGT eingeteilt:
- Vogt Typ I: Die obere Speiseröhre endet als Blindsack
- Vogt Typ II: Atresie ohne Verbindung zur Luftröhre
- Vogt Typ IIIa: Verbindung zur Luftröhre des oberen Segments, das untere Segment endet im Blindsack
- Vogt Typ IIIb: Verbindung zur Luftröhre des unteren Segments, das obere Segment endet im Blindsack
- Vogt Typ IIIc: Verbindung zur Luftröhre von oberen und unteren Segment
- Vogt Typ IV: Keine Atresie der Speiseröhre aber Verbindung zur Luftröhre (sog. "H-Fistel")
Die neugeborenen Patienten fallen durch Husten, Speicheln und Verschlechterung ihres Allgemeinzustandes auf. Bei Vogt IV leiden die erkrankten Säuglinge an wiederholten Aspirationspneumonien (Lungenentzündungen).
Diagnostik:
Vorgeburtlich weist ein Polyhydramnions (zu viel Fruchtwasser) im Ultraschall auf eine mögliche Ösophagusatresie hin. Ein federnder Stopp nach Sondierung der Speiseröhre ist hinweisend auf eine Ösophagusatresie. Magensaft kann nicht aspiriert werden. Eine Röntgenaufnahme des Brustkorbes zeigt die Luftfüllung des oberen Blindsackes (sog. Medaillonzeichen), und gegebenenfalls eine Luftfüllung des Darmes als Hinweis auf eine untere Fistel.
Behandlungsbedarf / konservative Behandlung:
Es besteht eine Operationsindikation.
Operation:
Die Operation werden beide Ösophagussegmente per End-zu-End-Anastomose miteinander verbunden, dies kann auch mit minimalinvasiven Operationsmethoden erfolgen. Eine bestehende Fistel zur Luftröhre wird verschlossen. Bei langstreckigen Atresien kommen Techniken zur Verlängerung der Speiseröhre zum Einsatz.
Ergebnisse:
Die Prognose ist abhängig von der Ausprägung der Ösophagusatresie, von weiteren Fehlbildungen, der frühzeitigen Therapie, sowie dem Reifegrad und dem Geburtsgewicht des Neugeborenen. Bei manchen Kindern entwickelt sich im Bereich der Anastomose eine Enge, die bougiert werden muss.
Häufige Fragen:
Kann mein Kind nachher alles essen? - Besteht keine Enge im Bereich der Speiseröhre kann das Kind alles essen.
Sind Kontrollen erforderlich? - Bei guter Gewichtentwicklung und Nahrungsaufnahme sind keine Kontrollen erforderlich.
Hat mein Kind Schmerzen beim Schlucken? - Schmerzen beim Schlucken sind nicht zu erwarten.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Duodenalatresie
Duodenalatresie
Eine Duodenalatresie ist eine seltene Erkankung des Neugeborenen, bei der die Nahrungspassage durch den Zwölffingerdarm nicht möglich ist. Entweder besteht eine Membran, die den Darm von innen verschliesst, oder der Darm wird von aussen z.B. durch eine ringförmig angelegte Bauchspeicheldrüse (Pankreas anulare) zusammengedrückt. In Greifswald wird eine Duodenalatresie meistens schon vor der Geburt durch das charakteristische „double-bubble“ Zeichen in der Feindiagnostik entdeckt. Die Schwangerschaft kann aber in der Regel in aller Ruhe beendet und die Geburt abgewartet werden.
Am ersten Lebenstag wird die Diagnose Duodenalatresie sonographisch bestätigt und der Mageninhalt durch eine Sonde abgeleitet, damit das Neugeborene nicht erbricht. Dann wird durch einen kleinen Schnitt am Bauch die Nahrungspassage hergestellt, indem die Darmanteile vor und nach der Duodenalatresie miteinander verbunden werden.
Die Magensonde wird dabei über die Anastomose geschoben, sodaß das Kind sehr bald schon mit Muttermilch ernährt werden kann. In der Regel dauert es nur wenige Tage, bis es dann selbst an der Brust zu trinken beginnt und nach Hause entlassen werden kann.
Während der ersten Wochen werden Kind und Eltern von unserem interdisziplinären Team des Zentrums für angeborene Fehlbildungen der Universitätsmedizin Greifswald betreut. Wenn es dann gut an Gewicht zunimmt, bleibt von der Operation selbst nur der kleine Schnitt am Bauch und das Kind kann ganz normal zum Kinderarzt gehen. Das Kind und später der Erwachsene kann alles essen, es gibt keine Einschränkungen.
Kontakt:
Klinik und Poliklinik für Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@uni-greifswald.de
Dünndarmatresie
Dünndarmatresie
Symptome:
Es handelt sich um angeborene Unterbrechungen der Darmkontinuität. In den Darmabschnitten oberhalb der Unterbrechung kommt es zum Aufstau von Darminhalt und damit in Abhängigkeit von der Höhe des Defektes früher oder später zum Erbrechen. Wird die Diagnose zu spät gestellt kann es zur Perforation des Darmes kommen. Die Folge ist eine schwere Entzündung des Bauchraums.
Diagnostik:
Die Diagnose wir häufig schon vor der Geburt im Ultraschall gestellt. Ein Polyhydramnion (zu viel Fruchtwasser) im vorgeburtlichen Ultraschall kann auf eine Darmatresie hinweisen. Nach der Geburt ist der Kostaufbau erschwert. Es kommt zu galligem Erbrechen je Höher die Stenose desto früher. Typisch ist eine fehlende oder verzögerte Mekonium (Neugeborenenstuhl) Passage. Bei tiefen Stenosen zeigt sich ein ausladender Bauch. In einer Sonographie des Bauches fallen die erweiterten Darmschlingen auf.
Behandlungsbedarf / konservative Behandlung:
Es besteht eine Operationsindikation.
Operation:
Bei der operativen Therapie wird der Bauchraum eröffnet und der Darm sorgfältig inspiziert um weitere Anomalien zu entdecken und ggf. zu behandeln. Der obere Darmanteil ist meist stark erweitert. Der untere nicht befahrene Darmanteil ist verkümmert und weißt ein kleines Darmlumen auf. Zur primären Verbindung beider Darmanteile ist eine schräge Anastomosentechnik notwendig.
Ergebnisse:
Bei einer zeitnahen Therapie besteht eine sehr gute Prognose. Die Darmtätigkeit kann für ein paar Tage nach der Operation erschwert sein.
Häufige Fragen:
Kann mein Kind alles essen? - Ja.
Sind Kontrollen erforderlich? - Bei guter Gewichtsentwicklung und Nahrungsaufnahme sind keine Kontrollen erforderlich.
Wie groß wird die Narbe? - Die Narbe erstreckt sich quer über den Bauch oberhalb des Nabels.
Kontakt:
Klinik und Poliklinik für Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@uni-greifswald.de
Dickdarmatresie
Dickdarmatresie
Symptome:
Es handelt sich um angeborene Unterbrechungen der Darmkontinuität. In den Darmabschnitten oberhalb der Unterbrechung kommt es zum Aufstau von Darminhalt und damit in Abhängigkeit von der Höhe des Defektes früher oder später zum Erbrechen. Wird die Diagnose zu spät gestellt kann es zur Perforation des Darmes kommen. Die Folge ist eine schwere Entzündung des Bauchraums.
Diagnostik:
Die Diagnose wir häufig schon vor der Geburt im Ultraschall gestellt. Ein Polyhydramnion (zu viel Fruchtwasser) im vorgeburtlichen Ultraschall kann auf eine Darmatresie hinweisen. Nach der Geburt ist der Kostaufbau erschwert. Es kommt zu galligem Erbrechen je Höher die Stenose desto früher. Typisch ist eine fehlende oder verzögerte Mekonium (Neugeborenenstuhl) Passage. Bei tiefen Stenosen zeigt sich ein ausladender Bauch. In einer Sonographie des Bauches fallen die erweiterten Darmschlingen auf.
Behandlungsbedarf / konservative Behandlung:
Es besteht eine Operationsindikation.
Operation:
Bei der operativen Therapie wird der Bauchraum eröffnet und der Darm sorgfältig inspiziert um weitere Anomalien zu entdecken und ggf. zu behandeln. Der obere Darmanteil ist meist stark erweitert. Der untere nicht befahrene Darmanteil ist verkümmert und weißt ein kleines Darmlumen auf. Zur primären Verbindung beider Darmanteile ist eine schräge Anastomosentechnik notwendig.
Ergebnisse:
Bei einer zeitnahen Therapie besteht eine sehr gute Prognose. Die Darmtätigkeit kann für ein paar Tage nach der Operation erschwert sein.
Häufige Fragen:
Kann mein Kind alles essen? - Ja.
Sind Kontrollen erforderlich? - Bei guter Gewichtsentwicklung und Nahrungsaufnahme sind keine Kontrollen erforderlich.
Wie groß wird die Narbe? - Die Narbe erstreckt sich quer über den Bauch oberhalb des Nabels.
Kontakt:
Klinik und Poliklinik für Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@uni-greifswald.de
Gastroschisis
Gastroschisis
Symptome:
Bei der Gastroschisis handelt es sich um einen Defekt der Bauchwand in der Mittellinie. Es resultiert ein offener Vorfall von Darmanteilen vor die Bauchwand. Im Unterschied zur Omphalozele ist der Darm nicht von der Nabelschnur geschützt und flotiert frei im Fruchtwasser.
Diagnostik:
Die Diagnose wird vorgeburtlich im Ultraschall gestellt. Nach der Geburt ist die Fehlbildung offensichtlich.
Behandlungsbedarf:
Es besteht eine Operationsindikation.
Operation:
Die operative Therapie besteht in der Rückverlagerung der vorgefallenen Darmanteile in die Bauchhöhle und dem Verschluss der Bauchwand. Bei einer kleinen Bauchhöhle und großen Anteilen vorgefallenen Darms können alternativ die Darmschlingen zunächst in einen sterilen Kunststoffbeutel verbracht werden. Dieser wird an der Bauchdecke fixiert und über dem Kind aufgehängt wird. In den folgenden Tagen gleiten die Darmschlingen dann entsprechend der Schwerkraft in den Bauchraum, die Lücke der Bauchwand wird im Anschluss verschlossen.
Ergebnisse:
Die Langzeitergebnisse sind gut und im Einzelnen Abhängig von der Größe des Bauchwanddefektes und vorliegenden Begleitfehlbildungen.
Häufige Fragen:
Muss mein Kind sofort operiert werden? - Die Operation wird erst nach Stabilisierung des Kindes auf der neonatologischen Intensivstation erfolgen.
Bleiben Spätfolgen zurück? - Spätfolgen an den Bauchorganen sind nicht zu erwarten.
Muss bei der Ernährung etwas beachtet werden? - Anfänglich kann die Nahrungsaufnahme erschwert sein, dann wird das Kind vorrübergehend auf der neonatologischen Intensivstation mit Infusionslösungen ernährt.
Kontakt:
Klinik und Poliklinik für Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@uni-greifswald.de
Omphalozele
Omphalozele
Symptome:
Eine Omphalozele ist ein Nabelschnurbruch und entsteht zwischen dem 32 und 70 Tag der Schwangerschaft. Während der Entwicklung des Kindes im Mutterleib werden die Bauchorgane nach außen vor die Bauchwand verlagert. Bei der Omphalozele bleibt die physiologische Rückverlagerung der Organe in den Bauchraum aus. Die Bauchorgane sind dabei von der Nabelschnur umgeben und bleiben dadurch geschützt.
Diagnostik:
Die Omphalozele wird im vorgeburtlichen Ultraschall diagnostiziert. Nach der Geburt ist die Fehlbildung offensichtlich.
Behandlungsbedarf / konservative Behandlung:
Die Therapie ist in der Regel operativ und besteht in einer Rückverlagerung des Bruchsackinhaltes in die Bauchhöhle und primärem Verschluss der Bauchdecken.
Operation:
Bei der Operation werden die Eingeweide in den Bauch zurückverlagert und der Bauch verschlossen. Bei sehr großen Brüchen und kleiner Bauchhöhle muss ein zweizeitiges Vorgehen gewählt werden. Im ersten Schritt erfolgt eine Teilreposition und ein Verschluss der Bauchdecke mit synthetischen Interponaten. Die Bauchdecken werden dann im Verlauf der nächsten Tage endgültig verschlossen.
Ergebnis:
Die Langzeitergebnisse sind sehr gut und im Einzelnen von vorliegenden Begleitfehlbildungen abhängig.
Häufige Fragen:
Muss mein Kind sofort operiert werden? - Die Operation wird erst nach Stabilisierung des Kindes auf der neonatologische Intensivstation erfolgen.
Bleiben Spätfolgen zurück? - Spätfolgen an den Bauchorganen sind nicht zu erwarten.
Muß bei der Ernährung etwas beachtet werden? - Anfänglich kann die Nahrungsaufnahme erschwert sein, dann wird das Kind vorübergehend auf der neonatologischen Intensivstation mit Infusionslösungen ernährt.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Zwerchfellhernie
Zwerchfellhernie
Symptome:
Bei einer Zwerchfellhernie besteht eine Lücke im Zwerchfell. Dadurch kommt es zur Verlagerung von Bauchorganen in die Brusthöhle. Die Lunge kann sich als Folge dessen nicht normal entwickeln. Nach der Geburt ist es der Lunge nicht möglich sich regelrecht zu entfalten. Die Kinder fallen nach der Geburt durch eine Atemnot auf.
Diagnostik:
Im vorgeburtlichen Ultraschall wird die Diagnose gestellt. Nach der Geburt sind in der Röntgenaufnahme des Thorax neben der Lunge die Bauchorgane wie z.B. der Darm sichtbar.
Behandlungsbedarf / konservative Behandlung:
Es besteht eine Operationsindikation.
Operation:
Bei der Operation werden die Organe aus dem Brustkorb in den Bauchraum zurückverlagert und die Lücke im Zwerchfell verschlossen, dies kann auch mit minimalinvasiven Operationsmethoden erfolgen. Ist die Lücke zu groß und kann nicht durch eine einfache Naht verschlossen werden, wird ein Kunststoffpatch in den Defekt eingenäht. Beim Zurückverlagern der Bauchorgane in die Bauchhöhle kann es vorkommen, dass diese nicht genug Platz bietet. In einem solchen Fall wird die Bauchhöhle ebenfalls mit synthetischen Interponaten verschlossen. Wachsen die Bauchdecken im Verlauf so kann das Fremdmaterial in einem weiteren Eingriff entfernt werden und ein kompletter Bauchdeckenverschluss erfolgen.
Ergebnisse:
Entscheidend für die Prognose ist das Ausmaß der Lungenfehlbildung und anderer begleitender Fehlbildungen. Die meisten Patienten erreichen später eine normale Lungenfunktion.
Häufige Fragen:
Muss mein Kind später auf etwas achten? - Nein.
Kann mein Kind Sport machen? - Bei einer normalen Lungenfunktion spricht nichts gegen sportliche Aktivität.
Wie groß ist die Narbe? - Die Narbe erstreckt sich über den linken Oberbauch unterhalb des Rippenbogens.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Analatresie
Analatresie
Was ist eine Rektum-/Analatresie?
Bei Anorektalen Fehlbildungen handelt es sich um Entwicklungsdefekte des Enddarms die sich in vielfältiger Weise äußern können. Der Anus kann bei blind-endendem Enddarm völlig fehlen, es kann aber auch zu Verbindungsgängen (Fisteln) zwischen dem Enddarm und der Haut des Dammes, der Genitale oder dem unteren Harntrakt kommen. Bei den sogenannten Kloakenfehlbildungen des weiblichen Geschlechtes findet sich eine gemeinsame Mündung des Darmes, der Vagina und der Harnröhre/ Blase.
Die Erkrankung betrifft etwa 1:3000 – 1:4000 Neugeborene, dabei sind Jungen etwas häufiger betroffen. Die Erkrankung kann alleine oder in Kombination mit anderen Fehlbildungen auftreten.
Wie wird eine Rektum-/Analatresie diagnostiziert?
Die Rektum-/Analatresie wird in der Regel bei der körperlichen Erstuntersuchung des Neugeborenen nach der Geburt (U1) festgestellt. Bei völligem Fehlen des Rektums/Anus ist eine Ultraschalluntersuchung zur Bestimmung der Höhe des blind endenden Enddarms erforderlich. Eine Röntgenuntersuchung des Bauchraums in Kopftieflage nach ca. 16-24 Lebenstunden gibt ebenfalls Auskunft über die Höhe des Blindsacks. Durch die körperliche Untersuchung ihres Kindes, durch Ultraschalluntersuchungen der Nieren, der Wirbelsäule und des Herzens werden weitere Fehlbildungen ausgeschlossen.
Wie wird die Rektum-/Analatresie behandelt?
Wird ihr Kind mit einer Rektum-/Analatresie geboren, werden wir in der Universitätsmedizin Greifswald die individuell sinnvolle Therapie mit ihnen besprechen. Diese hängt maßgeblich von der genauen Ausprägung der Fehlbildung abhängt.
Bei ausreichend breiten äußeren Fisteln ist eine Stuhlentleerung über die Fistel möglich, eventuell kann eine Aufweitung der Fistel (Bougierung) erforderlich sein. Bei blind endendem Enddarm kommt es zu einem Darmverschluß. Abhängig von der Höhe des Blindsacks ist es erforderlich den Darm zunächst durch einen künstlichen Dickdarmafter (Kolostoma) zu entlasten oder bei geringem Abstand des Enddarms zur Haut frühzeitig eine plastische Rekonstruktion des Afters (Anorektoplastik) am 2. -3. Lebenstag durchzuführen.
Wird ein Kolostoma angelegt erfolgt im Verlauf eine Darstellung des Darms mittels Kontrastmittelröntgen um kleine Verbindungsgänge ( Fisteln) zur Vagina, Blase oder den Harnwegen auszuschließen. Die Rekonstruktion (Posteriore sagitale Anorektoplastik nach Peña - PSARP) erfolgt dann ca. 4-6 Monate später.
Etwa 2 Wochen nach der Anorektoplastik erfolgt die Aufdehnung des neugeschaffenen Afters mittels speziellen Hegarstiften. Die Eltern werden darin angeleitet und führen diese Aufdehnung dann 2 x täglich zuhause durch. Ist der After ausreichend weit kann ca. 4-6 Wochen nach der Anorektoplastik die Rückverlagerung des Kolostomas erfolgen.
Welche Komplikationen gibt es? Was für Kontrollen sind erforderlich?
Es kann zu Verengung des Afters (Analstenose) und der Vagina (Vaginalstenose) kommen. Im Afterbereich kann es zum Vorfall der Schleimhaut oder des Enddarms kommen (Mukosa-/ Rektumprolaps). Kinder mit tiefer Analatresie leiden häufiger unter chronischer Obstipation (Verstopfung) und Kinder mit hohen Atresieformen eher unter Stuhlschmieren und Inkontinenz, wobei Kombinationen möglich sind.
Die postoperativen Ergebnisse der anorektalen Malformationen hängen sehr von der Höhe der Fehlbildung und der Anlage des muskulären Beckenbodens ab. Sehr tief gelegene Atresien haben dabei deutlich bessere Ergebnisse als hohe Fisteln z.B. zur Prostata oder Kloakenfehlbildungen.
Wir in der Universitätsmedizin Greifswald begleiten sie in der Nachsorge bis zur Pubertät und darüber hinaus um eventuelle Komplikationen frühzeitig erkennen und behandeln zu können.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Thoraxchirurgie
Trichterbrust
Trichterbrust
Was ist eine Trichterbrust?
Die Trichterbrust (Pectus excavatum) ist eine Deformität der vorderen Brustwand, bei der es durch Fehlentwicklung des Rippenknorpels am Übergang zum Brustbein zu einer Einsenkung des Brustbeins kommt. Die Einsenkung kann dabei symmetrisch oder asymmetrisch sein. Häufig kommt es zusätzlich zum Vorstehen der unteren Rippenbögen (sog. Rib flaring). Jungen sind deutlich häufiger betroffen als Mädchen.
In vielen Fällen ist die Deformität angeboren, bei anderen tritt sie erst später auf. Meist verstärkt sich die Einsenkung des Brustbeines rasch im Rahmen des pubertären Wachstumsschubes. Eine individuelle Prognose ist schwierig, eine spontane Rückbildung jedoch nicht zu erwarten.
Mögliche Begleiterkrankungen können z.B. die Skoliose oder ein Marfan-Syndrom sein.
Welche Symptome können bei einer Trichterbrust auftreten?
Häufig berichten die Patienten über eine subjektiv eingeschränkte körperliche Belastbarkeit bis hin zu Luftnot. Auch Schmerzen im Brustkorb, Herzrasen oder Schwindel können auftreten. Meist kommt es insbesondere in der Pubertät zu Störungen des Körperschemas und des Selbstwertgefühls durch die Deformität. Dies kann zu Vermeidungsverhalten (z. B. bei sportlichen Aktivitäten wie Schwimmen etc.) bis hin zum sozialen Rückzug der Kinder/Jugendlichen führen und damit die psychosoziale Entwicklung ernsthaft gefährden.
Wie wird die Diagnose gestellt?
Die Trichterbrust ist zunächst eine Blickdiagnose (d.h. die Diagnose wird über die typische Form des Brustkorbs gestellt). Um den Schweregrad und die Symptome weiter abzuklären sind jedoch (angepasst an das Alter und die Beschwerden) noch folgende Untersuchungen notwendig
- MRT des Brustkorbs
Bei dieser Schnittbildgebung, die im Gegensatz zum CT ohne ionisierende Strahlung auskommt, können die Brustwand und die im Brustkorb gelegenen Organe beurteilt werden. Zur Bestimmung des Schweregrades der Trichterbrust wird der Haller-Index (HI) bestimmt (Quotient aus innerem Querdurchmesser des Brustkorbs und kleinstem Abstand zwischen Wirbelvorderkannte und Brustbein-Hinterkante). Beim Gesunden beträgt dieser Index ca. 2,5-2,7. Von einer milden Trichterbrust spricht man bei einem Haller-Index von < 3,5, von einer moderaten Trichterbrust bei einem HI von 3,5-4,5 und von einer schweren Trichterbrust bei einem HI von > 4,5. Weiterhin kann mittels der MRTdie Verkippung des Brustbeines sowie eine mögliche Kompression oder Verlagerung von Herz und Lungen durch die Einziehung des Brustbeines beurteilt werden.
- Lungenfunktionsuntersuchung
Hierbei wird gemessen ob die Lunge eine normale Kapazität hat oder ob es Störungen bei der Atmung gibt. Bei Kindern und Jugendlichen findet sich häufig auch bei schwerer Trichterbrust keine messbare Einschränkung der Lungenfunktion. Am häufigsten werden restriktive oder obstruktive Ventilationsstörungen beobachtet.
- Echokardiographie
Bei dieser Untersuchung wird das Herz mittels Ultraschalles untersucht. Die Lage des Herzens und eine eventuelle Kompression durch die Einziehung des Brustbeins können dargestellt werden, außerdem wird die Funktion des Herzens beurteilt. Wenn Einschränkungen vorliegen betrifft dies meist die rechte Herzhälfte.
- Kinderpsychologische Untersuchung
Bei dieser Untersuchung wird mittels spezieller Fragebögen sowie im Gespräch mit dem Patienten und den Eltern erfasst, ob der Patient unter der Brustkorbdeformität psychisch leidet, ob er ggf. sogar gefährdet ist, sozioemotionale Entwicklungsstörungen oder soziales Vermeidungsverhalten auszubilden. Insbesondere bei einer geplanten operativen Therapie wird auch geprüft, ob eine Einsichtsfähigkeit in den Eingriff vorliegt.
Welche Therapien gibt es?
Die mögliche Therapie richtet sich immer nach dem Alter des Kindes, der Ausprägung der Trichterbrust und den individuellen Beschwerden.
- Physiotherapie
Das Trainieren der Brust-, Bauch- und Rückenmuskulatur kann insbesondere die begleitende Fehlhaltung beseitigen und den Stützapparat kräftigen. Dies ist auch für alle weiteren Therapien hilfreich. Die bei der Krankengymnastik erlernten Übungen sollten konsequent auch zuhause weitergeführt werden um ein dauerhaft gutes Ergebnis zu erzielen.
Des Weiteren empfehlen wir Ausdauersportarten (z.B. Schwimmen, Leichtathletik etc.)
- Saugglocke
Die „Saugglocke nach Eckardt Klobe zur nicht-invasiven Anhebung der Trichterbrust“ besteht aus medizinischem Silikon. Es gibt verschiedene Formen je nach Größe des Patienten und Form der Brustwandeinziehung. Mittels einer Handpumpe wird ein Unterdruck angelegt, welcher das Brustbein nach außen zieht. Bei konsequenter Anwendung (2 x täglich 30 min oder länger) kann es so zu einer dauerhaften Anhebung des Brustbeins kommen.
- Operative Therapie
Die minimalinvasive Trichterbrustkorrektur (MIRPE: minimally invasive repair of pectus excavatum) n. D. Nuss (1998) ist mittlerweile der Goldstandard der operativen Therapie der Trichterbrust. Dabei wird unter Kamerasicht (Thorakoskopie) ein individuell angeformter Metallbügel (Pectusbar) hinter dem Brustbein an der tiefsten Stelle der Einsenkung entlanggeführt. Durch das Drehen des Bügels und die Fixierung dieses mittels Halteplatten an der vorderen oder seitlichen Brustwand kommt es zu einer unmittelbaren Anhebung des Brustbeins.
Nicht immer lassen sich Asymmetrien oder das Vorstehen der unteren Rippenbögen mit dieser Methode vollständig beseitigen.
Der ideale OP-Zeitpunkt ist bei Jugendlichen gegen Ende des Wachstumsabschlusses (ca. 13.-17. Lebensjahr)
Für den Eingriff ist ein stationärer Aufenthalt von ca. einer Woche erforderlich. Aufgrund der raschen Änderung der Brustkorbform und der Rückstellkräfte des Brustkorbs treten initial immer Schmerzen auf. Für die Phase unmittelbar nach der Operation wird durch unsere Narkoseärzte ein Rückenmarks-naher Schmerzkatheter (thorakaler PDK) angelegt über den eine kontinuierliche Schmerzmedikation verabreicht wird. Bei Bedarf wird diese mit einer durch den Patienten steuerbaren Medikamentenpumpe ergänzt. Zusätzlich werden intravenöse und später orale Schmerzmittel gegeben, mit denen die Patienten auch nach Hause entlassen werden können.
In den ersten 6 Wochen nach der Operation sollte keinerlei sportliche Betätigung erfolgen. Nach der 6. Woche kann mit Physiotherapie sowie leichten Ausdauerbelastungen (z.B. Schwimmen, Joggen) begonnen werden. Rotationsbewegungen und schweres Heben oder Tragen sollten für 12 Wochen gemieden werden. Nach 12 Wochen kann eine sportliche Betätigung wieder aufgenommen werden. Von Vollkontakt-Sportarten (z.B. Kampfsport, Eishockey, Football etc.) raten wir ab.
Die operative Entfernung des Bügels erfolgt nach ca. 2-3 Jahren. Bis dahin hat der Brustkorb Zeit sich derart umzuformen, dass meist eine bleibende Aufrichtung des Brustbeins erzielt wird.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Lungensequester
Lungensequester
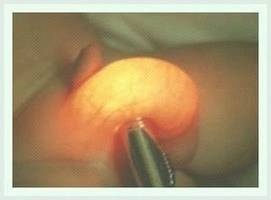
Synonym: bronchopulmonaler Sequester
Als Lungensequester (LS) wird ein funktionsloser Lungenanteil bezeichnet, der nicht am Gasaustausch der Lunge teilnimmt. Diese Fehlbildung ist angeboren und macht 0,2-0,6% aller angeborenen Fehlbildungen aus.
Es gibt zwei Arten von LS, wobei die extralobären bei Säuglingen und Kleinkindern häufiger sind. Sie werden meist vor der Geburt (pränatal) im Routineultraschall erkannt.
Angeborene LS sind in den meisten Fällen unproblematisch für das Neugeborene und führen nur selten zu Beeinträchtigungen. Eine chirurgische Therapie in Form einer Entfernung der Fehlbildung ist dementsprechend nur selten notwendig und wird meist gut vertragen.
Die Prognose ist abhängig von Begleiterkrankungen und in der Regel gut.
Literatur:
- Cooke CR: Bronchopulmonary sequestration. Respir Care 2006; 51: 661-664
- Gluer S, v Schweinitz D: Fehlbildungen und Erkrankungen der Lunge, der Pleura und des Mediastinums. In v Schweinitz D, Ure B: Kinderchirurgie. Springer 2009
- Salmons S: Pulmonary sequestration. Neonatal Netw 2000; 19: 27-31
- Savic B, Birtel Fj, Tholen W, Funke HD, Knoche R: Lung sequestration: report of seven cases and review of 540 published cases. Thorax 1979; 34:96-101
- Qian X, Sun Y, Liu D, Wu X, Wang Z, Tang Y: Pulmonary sequestration: a case report and literature review. Int J Cin Exp Med 2015; 8: 21822-21825
- Wei Y, Li F: Pulmonary sequestration: a retrospective analysis of 2625 cases in China. Eur J Cardiothorac Surg 2011; 40: e39-e42
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Lungenemphysem
Lungenemphysem
Kongenitales lobäres Emphysem
Das lobäre Emphysem ist die angeborene Überblähung eines Lungenlappens.
Diese Erkrankung betrifft meist nur einen Lungenlappen. Manchmal läßt sich dies bereits in der vorgeburtlichen Routine - Ultraschalluntersuchung vermuten. Es gibt aber auch noch andere angeborene Lungenerkrankungen, die dem lobären Emphysem im Ultraschall ähneln.
Als Ursache können Knorpelstrukturen, Lungenfehlbildungen, atypische Gefäßverläufe oder Schleimpfropfen gefunden werden; in der Hälfte der Fälle findet man jedoch keine Ursache.
Es können begleitende Herzfehlbildungen vorhanden sein.
Betroffene Neugeborene werden operiert - der Zeitpunkt hängt davon ab, wie es dem Kind nach der Geburt geht. In der Operation wird der betroffene Lungenanteil entfernt.
Die Prognose ist abhängig von einer Begleiterkrankung und in der Regel sehr gut.
Literatur:
- Gluer S, v Schweinitz D: Fehlbildungen und Erkrankungen der Lunge, der Pleura und des Mediastinums. In v Schweinitz D, Ure B: Kinderchirurgie. Springer 2009
- Pinkerton HJ, Oldham KT: Lung. In: Oldham KT, Colombani PM, Foglia RP, Skinner MA (eds): Principles and practice of pediatric surgery. Lippincott Williams & Wilkins, Philadelphia 2005; 951-982
- Sylvester KG, Albanese MD: Bronchopulmonary malformations. In: Ashcraft KW, Holcomb GW, Murphy JP (eds.): Pediatric Surgery. Elsevier Saunders, Philadelphia 2005; 276-289
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
zystich-adenomatoide Malformation
zystich-adenomatoide Malformation
englisch: congenital cystic adenomatoid malformation (CCAM)
Die zystische adenomatoide Malformation der Lunge ist eine angeborene Fehlbildung. Sie kommt bei 1:11000 bis 1:35000 Lebendgeborenen vor, ist also sehr selten.
Diese Erkrankung betrifft meist nur einen Lungenlappen, in 60% der Fälle den linken. Meistens fällt dies bereits im der vorgeburtlichen Routine - Ultraschalluntersuchung auf.
Es gibt eine Klassifikation (nach Stocker), deren Einteilung auf äußerer Erscheinungsform der Zystenstruktur (Zyste = Hohlraum) und bindegeweblichen Strukturen im Mikroskop beruht.
Typ I, makrozystisch: häufig; eine oder mehrere luftgefüllte Zysten >2cm Durchmesser
Typ II, mikrozystisch: häufig; mehrere, teilweise verbundene Zysten bis 1cm Durchmesser
Typ III, solide: selten; luftleere unreife Strukturen
Typ IV, Typ 0: sehr selten
Betroffene Neugeborene können unauffällig sein und verschiedene Formen der Atemnot aufweisen. Kinder mit klinischen Symptomen werden nach Stabilisierung operiert. Hierbei wird der betroffene Lungenanteil entfernt.
Ob und zu welchem Zeitpunkt ein symptomloses Kind operiert werden sollte, ist Gegenstand aktueller Diskussion und muß im Einzelfall entschieden werden.
Die Prognose ist abhängig von Begleiterkrankungen und in der Regel gut.
Literatur
- Gluer S, v Schweinitz D: Fehlbildungen und Erkrankungen der Lunge, der Pleura und des Mediastinums. In v Schweinitz D, Ure B: Kinderchirurgie. Springer 2009
- Granata C, Gambini C, Balducci T, TomaP, Michelazzi A, Conte M, Jasonni V: Bronchoalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol 1998; 25: 62-66
- MacSweeny F, Papagiannopoulos K, Goldstraw P, Shappard MN, Corrin B, Nicholson AG: An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol 2003; 27: 1139-1146
- Sauvat F, Michel JL, Benachi A, Edmomd S, Revillon Y: Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg 2003; 38: 548-552
- Wilson RD, Hedrick HL, Liechty KW, Flake AW, Johnson MP, Bebbington M, Adzick NS: Cystic adenomatoid malformation of the lung: review of genetics, prenatal diagnosis and in utero treatment. Am J Med Genet A 2006; 140: 151-155
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Abdominalchirurgie
Gastroösophagealer Reflux
Gastroösophagealer Reflux
Definition:
Unter diesem Begriff versteht man zunächst einmal nur das Zurücklaufen von Mageninhalt in die Speiseröhre. Dies geschieht im Säuglingsalter bei fast allen Kindern gelegentlich. Zu einer Krankheit wird der Reflux erst, wenn das Kind unter den Folgen des Reflux leidet.
Symptome:
Das meist als erstes auffallendes Symptom eines gastroösophagealen Reflux ist das häufige Spucken oder Erbrechen. Da im Säuglingsalter die Speiseröhre noch sehr dünn ist, tritt der zurückfließende Mageninhalt aus dem Mund wieder nach außen. Wie bereits gesagt, ist dieses in einem gewissen Rahmen völlig normal, da die Speiseröhre noch sehr flach in den Magen mündet. Erst weitere Symptome lassen den Reflux krankhaft werden. Zudem gibt es viele weitere Gründe und Krankheiten warum ein Kind häufig erbricht, die abgeklärt werden müssen. Es gibt auch kleine Patienten, die unter einem Reflux leiden, ohne zu erbrechen, da der Mageninhalt nicht bis in den Mund läuft.
Die zurückfließende Magensäure verursacht bei den Kindern Schmerzen und Sodbrennen, darum leiden Sie unter Schlafstörungen und schreien viel, zeigen ein Anspannen von Händen und Füßen und haben ggf. Schweißausbrüche.
Der Mageninhalt kann, ohne aus dem Mund abzufließen, auch in die Luftröhre gelangen. Dies führt zu Husten und röchelnden Geräuschen bei der Atmung. Bei immer wieder auftretenden Infektionen der Lunge und chronischen Husten sollte daher auch an einen Reflux gedacht werden.
Die Nahrung wird immer wieder erbrochen und durch die Schmerzen, die das Kind beim Füttern hat, kommt es zur Nahrungsverweigerung und Trinkschwäche. Besonders im Säuglingsalter kann dies zu mangelnder Gewichtszunahme und Gedeihstörungen führen.
Der saure Magensaft kann außerdem die empfindlichen Milchzähne schädigen.
Diagnostik:
Im Säuglingsalter leidet 1 Kind von 500 unter der Symptomatik, unter Kleinkindern etwa noch 1 von 2000 Kindern.
Da also der Reflux selbst nicht immer eine Krankheit darstellt und sich im Laufe der Entwicklung zurückbilden kann, bedarf es einer genauen und sorgfältigen Diagnostik. So lassen sich die Fälle, bei denen man eine intensive Behandlung anwenden muss, von den anderen unterscheiden.
- 24 h pH-Metrie: Bei dieser Untersuchung wird ein sehr dünnes Kabel mit 2 Messsensoren durch die Nase in die Speiseröhre des Kindes gelegt. Ein Gerät zeichnet den pH-Wert, also den Säuregehalt, an den Messpunkten auf. Fließt nun saurer Magensaft in die Speiseröhre, verändert sich der pH-Wert und man kann sehen wann, wie lange, wie häufig und auch wie hoch der Magensaft aufgestiegen ist. Mit dieser Methode kann man jedoch keine Aussage zur Schädigung der Schleimhaut der Speiseröhre oder der Lunge treffen. Zudem ist die Messmethode sehr störanfällig.
- Röntgen-Brei-Schluck-Untersuchung: Diese Untersuchung ist nicht direkt dazu da, um einen Reflux nachzuweisen, sondern mit ihrer Hilfe kann man Fehlbildungen oder Erkrankungen, die einen Reflux verursachen, darstellen. Hierzu muss das Kind einen Brei schlucken, der nicht für Röntgenstrahlen durchlässig ist. In den anschließend angefertigten Röntgenbildern kann man dann sehen, wohin und wie schnell der Röntgenbrei fließt. Dadurch werden z.B. ein im Brustkorb liegender Magen oder eine Entleerungsstörung sichtbar.
- Gastroskopie: Die Magenspiegelung wird bei Kindern in Narkose durchgeführt. Dabei wird ein flexibler Schlauch, an dem eine Kamera angeschlossen ist, in die Speiseröhre und in den Magen eingeführt. So kann man z.B sehen, ob bereits Schäden an der Schleimhaut der Speiseröhre aufgetreten sind. Wenn dies der Fall ist, kann man mit einer kleinen Zange kleine Proben aus der Schleimhaut entnehmen und untersuchen.
- Bronchoskopie: Bei dieser Untersuchung schaut man sich mit einer Kamera die Luftröhre und ihre Aufzweigungen an. Dabei kann man feststellen, ob die aufsteigende Magensäure die Schleimhaut der Bronchien geschädigt hat. Auch bei dieser Untersuchung kann man kleine Proben für die Untersuchung im Labor entnehmen.
Behandlungsbedarf:
- Allgemeine Maßnahmen: Sollte festgestellt werden, dass ein Kind einer Behandlung bedarf, können oftmals schon einfache Maßnahmen die Beschwerden lindern. So kann das Andicken der Nahrung mit speziellen Präparaten Besserung bringen. Zudem sollten kleinere, aber dafür mehrere Mahlzeiten gefüttert werden. Die Mahlzeiten sollten nicht direkt vor dem Schlafengehen gegeben werden und das Kind sollte mit leicht erhöhtem Oberkörper schlafen. Zudem sollte auf das Rauchen in der Umgebung des Kindes verzichtet werden.
- Medikamentöse Therapie: Etwa bei 2/3 aller Kinder heilt der Reflux komplikationslos und ohne Eingriff ab. Bei diesen Kindern werden ausschließlich Medikamente verabreicht, mit denen die Säureproduktion im Magen gedämpft wird. Hierdurch können durch die Säure verursachte Entzündungen der Speiseröhre abheilen und es kommt zu keiner weiteren Zerstörung der Schleimhaut. Es werden hierzu auf der einen Seite Medikamente verwendet, die den Säuretransport in den Magen hemmen, die sogenannten Protonen-Pumpen-Hemmer, auf der anderen Seite Medikamente die bestimmte Rezeptoren blockieren, um die Säureproduktion zu senken, die sogenannten H2-Rezeptor-Antagonisten.
Operation:
Sollte sicht trotz medikamentöser Therapie und genügend langer Beobachtungszeit keine Besserung der Erkrankung einstellen und das Kind erheblich unter der Erkrankung leiden, sollte durch einen chirurgischen Eingriff eine Korrektur der vorhandenen anatomischen Gegebenheiten vorgenommen werden. Im Gegensatz zur medikamentösen Therapie werden hierbei nicht nur die Symptome, sondern direkt die Ursache behandelt. So wird durch das manschettenartige Umschlingen des unteren Speiseröhrenendes mit Teilen des Magens ein Ventilmechanismus erzeugt. Ist der Magen gefüllt, spannt sich die Manschette um die Speiseröhre an und drückt so die Speiseröhre zu. Es kann kein Mageninhalt in die Speiseröhre fließen. Entleert sich der Magen, entspannt sich die Manschette und Nahrung kann wieder normal in den Magen gelangen.
Prinzipiell muss man 2 Operationstechniken unterscheiden: die offene und die laparoskopische Technik.
- offene Technik: Beim offenen Verfahren wird ein Schnitt direkt über den Magen gemacht. Diese Technik hat den Vorteil, dass der Operateur den gesamten Operationsbereich gut einsehen kann und auf Komplikationen rasch reagieren kann. Zudem ist die Operationsdauer hier geringer. Nachteile sind eine größere Narbe. Zudem braucht das Kind länger, um nach der Operation wieder auf die Beine zu kommen.
- laparoskopische Technik: Bei der laparoskopischen Technik wird im Prinzip der gleiche Eingriff über mehrere kleine Zugänge im Bauchraum durchgeführt. Dazu wird der Bauch zunächst aufgeblasen, um Platz zu schaffen. Dann wird eine Kamera eingeführt, über die der Operateur die Operation verfolgen kann. Über mehrere kleine Einschnitte werden dann die Instrumente, wie Scheren, Pinzetten und Skalpelle, an langen Stäben in den Bauch eingeführt und die Operation praktisch unter der Bauchdecke durchgeführt. Der Vorteil: da keine großen Wunden entstehen, gibt es keine großen Narben und der Patient hat weniger Schmerzen und ist schneller wieder auf den Beinen. Gelegendlich machen es Umstände jedoch notwendig offen zu operieren, oder eine zunächst laparoskopisch begonnene Operation offen fortzusetzen.
Bei beiden Techniken ist ein mehrere Tage andauernder stationärer Aufenthalt im Krankenhaus notwendig. Wenige Stunden nach der Operation kann das Kind meistens bereits wieder erste Nahrung zu sich nehmen.
Ergebnisse:
Bei den Erfolgsquoten unterscheiden sich die beiden Verfahren kaum, sie liegen bei jeweils etwa 90%. Etwa 4 Monate nach der OP wird noch einmal eine umfassende Diagnostik durchgeführt, um den Erfolg zu überprüfen.
Häufige Fragen:
Mein Kind spuckt häufig, ist es krank? - Nicht unbedingt. In den ersten Lebensmonaten ist das Ausspucken von Nahrung noch völlig normal, da der Einmündungswinkel der Speiseröhre in den Magen noch sehr flach ist und auch das Schlucken gelernt werden muss. Sollten aber Symptome wie Schmerzen, Schlafstörungen oder häufige Infekte der Atemwege auftreten, fragen Sie Ihren Kinderarzt danach.
Können die oben beschriebenen Symptome nur durch einen gastroösophagealen Reflux bedingt sein? - Nein. Viele andere Erkrankungen oder Fehlbildungen machen sehr ähnliche Symptome. So kann häufiges Erbrechen auch durch eine Engstelle am Magenausgang oder am Zwölffingerdarm verursacht werden. Zudem können Teile des Magens durch das Zwerchfell rutschen und ebenfalls diese Symptome auslösen. Deshalb ist eine genaue Untersuchung und Diagnostik sehr wichtig.
Wer behandelt mein Kind? - Erster Anlaufpartner sollte für Sie immer Ihr Kinderarzt sein. Er kennt Ihr Kind genau und wird weitere Schritte einleiten. Sollte er weitere Untersuchungen für nötig halten, werden diese in unserem Hause durch die Fachärzte der Kinderklinik durchgeführt. Wenn diese eine Operation für nötig erachten, werden sie die Kinderchirurgen hinzuziehen. Da sich an der Universitätsmedizin Greifswald alle Stationen unter einem Dach befinden, steht Ihnen also immer ein interdisziplinäres Team zur Verfügung und Sie bekommen immer die Betreuung, die Sie brauchen, ohne erst das Krankenhaus wechseln zu müssen.
Muss ich unbedingt ins Krankenhaus? - Meistens ja, denn viele Untersuchungen und Operationen lassen sich nur unter stationärer Überwachung durchführen. Wir versuchen jedoch soviel wie möglich im Vorfeld über unsere Ambulanz abzuklären. Zudem sind wir immer bemüht, Ihnen und vorallem Ihrem Kind den Aufenthalt so angenehm und so kurz wie möglich zu gestalten.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Endometriose
Endometriose
Als Endometriose wird das Vorkommen endometriumartiger Zellverbände(Schleimhaut, welche das Innere der Gebärmutter auskleidet) außerhalb der Gebärmutter bezeichnet.
Histopathologisch ist es eine gutartige Erkrankung welche infiltrativ wächst und viele Organe betreffen kann.
Es müssen nicht nur jugendliche Mädchen betroffen sein, in seltenen Fällen können auch junge Mädchen vor ihrer ersten Regelblutung betroffen sein
Die Ursache dieser Erkrankung ist trotz einiger Theorien noch unbekannt.
Symptome können immer wiederkehrende Unterbauchschmerzen, lange und starke Regelblutungen, Schmierblutungen sowie starke Menstruationsschmerzen sein.
Neben den möglichen medikamentösen Therapieoptionen ist auch die operative Sanierung der Herde durchführbar.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Ovarialzysten
Ovarialzysten
Ovarialzysten können sowohl bei Neugeborenen als auch bei Kleinkindern und Jugendlichen auftreten und sind häufig sonographische Zufallsbefunde.
Bei Neugeborenen nimmt man an, dass diese Ovarialzysten Folge der mütterlichen Hormonstimulation während der Schwangerschaft sind.
Funktionelle Ovarialzysten können häufig Verursacher von Menstruationsbeschwerden, Bauchschmerzen oder Miktionsbeschwerden sein.
Ab einer bestimmten Größe können diese Zysten zu einer Verdrehung des Ovars führen, die sich durch akut einsetzende Bauchschmerzen gelegentlich mit Erbrechen einhergehend zu Tage treten kann. Fälschlicherweise können solche Symptome als eine Blinddarmentzündung gedeutet werden. Eine Verdrehung des Ovars muss schnellst möglich operativ behoben werden, da diese zu einem Absterben des betroffenen Ovars führen kann.
Auf Grund dessen sind Ovarialzysten in regelmäßigen Abständen sonographisch zu kontrollieren.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Choledochuszyste
Choledochuszyste
Eine Choldedochuszyste ist eine kongenitale cystische Erweiterung des Ductus choledochus. Es wurde erstmals 1852 von Douglas beschrieben. Choledochuszysten sind sehr selten. Die Häufigkeit ist 1: 100 -200.000.
Die Ätiologie ist unklar. Es gibt verschiedene Theorien. Eine berühmte Theorie ist die "Common channel"- Theorie. In dieser Theorie handelt es sich um einen intrauterinen pancreatico-choledochalen Reflux, bei dem Pankreassekret und Galle in den Choledochus zurückläuft und eine Cholangitis hervorruft. In diesem Zusammenhang entsteht entweder eine Stenose, welche zur eine primären Dilatation oder zur eine prestenotische Dilatation des Choledochus führt.
Die Klinische Symptomatik ist vielfältig. Entweder besteht eine sogenannte typische "Trias Symptomatik":
- Bauchschmerzen
- Ikterus
- Tastbare Raumforderung im Oberbauch
Oder es wird später durch assoziierte Komplikationen klinisch symptomatisch:
- Cholangitis
- Choledocholithiasis
- Hepatolithiasis
- Pankreatitis
- Portale Hypertension
- Maligne Entartung
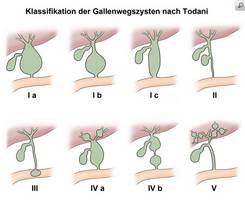
Einteilung
- Typ I:
Zystenbildung im Bereich des Ductus choledochus/hepaticus communis - Typ Ia:
Diffuse Erweiterung des Ductus choledochus - Typ Ib:
Segmentale Erweiterung des Ductus choledochus - Typ Ic:
Diffuse/zylindrische Erweiterung des Ductus choledochus/hepaticus communis - Typ II:
Divertikelartige Erweiterung des Ductus choledochus - Typ III:
Divertikelartige Erweiterung des Ductus choledochus innerhalb der Duodenalwand - Typ IV:
Kombination Typ I mit Erweiterung intrahepatischer Gallenwege Carroli
Die Diagnose wird mit Sonografie festgestellt. Es kann ein pränatal oder postnatal erweiteter Choldechus dargestellt werden. Bei dem sonografischen Verdacht wird ein MRCP durchgeführt. Der Vorteil von einem MRCP: es ist eine nicht invasive Untersuchung. Die andere mögliche Untersuchung ist ein ERCP. Der Nachteil dieser Untersuchung ist, dass es eine invasive Untersuchung ist und es kann postkonvertionelle aszendierende Infektion auftreten. Der Vorteil ist, dass man bei Diagnosestellung therapeutisch eine Papillotomie durchgeführen kann.
Therapie
- Typ I:
Radikale Resektion inkl. GB und Hepatikoduodenostomie/Hepatikojejunostomie mit Roux-Y Anastamose - Typ II:
Exzision des Divertikels - Typ III:
Bei Choledochozellen < 3cm- endoskopische Papillotomie
Bei Choledochozellen > 3cm- transduodenale Exzision der Zyste - Typ IV:
Bei Beschränkung der Zysten auf den linken Leberlappen- Hemihepatektomie
Sonst:
Exzision der exstrahepatischen Zysten mit Hepatikojejunostomie - Typ V (Carroli):
Hemihepatektomie bei unilobulärem Typ
Passagere Drainage und Lebertranplantatıon beı diffusen Typ
Morbus Hirschsprung
Morbus Hirschsprung
Was ist der Morbus Hirschsprung?
Der Morbus Hirschsprung (Synonym: Aganglionose/ HSCR) ist eine angeborene Fehlbildung des den Darm versorgenden Nervensystems. Bereits in der 5. – 12. Schwangerschaftswoche unterbleibt dabei das Einwandern der Nervenzellen in die Darmwand, genauer in den Plexus submucosus (Meißner) und des Plexus myentericus (Auerbach). Da dieser Prozess von oben nach unten stattfindet ist immer ein unterschiedlich langes Segment vom Enddarm nach oben hin betroffen. Durch die fehlenden Nervenzellen fehlt eine zum Enddarm hin gerichtete Darmbewegung.
Man unterscheidet je nach Länge des betroffenen Darmabschnittes:
- Die ultrakurze Aganglionose, welche auf den untersten Teil des Enddarms unterhalb des Beckenbodens begrenzt ist.
- Die kurzstreckige Aganglionose, welche das Sigma und den Enddarm betrifft (ca. 80- 85 % der Fälle)
- Die langstreckige Aganglionose – welche fast den gesamten Dickdarm (Zülzer-Wilson-Syndron, ca. 5%) betreffen kann
- Eine Aganglionose des gesamten Magen-Darm-Traktes tritt in 1% der Fälle auf
Die Erkrankung tritt in Mitteleuropa bei 1: 5000 Neugeborenen auf, dabei sind Jungen 4-mal häufiger betroffen als Mädchen. In 20% findet sich eine sporadische familiäre Häufung. Patienten mit Trisomie 21 haben ein 5%iges Risiko für einen Morbus Hirschsprung
In der Neugeborenenperiode fallen 90 % durch verspäteten Abgang des ersten Stuhlgangs (Mekonium), geblähten Bauch, Erbrechen und eventuell eine Darmentzündung (Enterokolitis) auf. 10% der Kinder zeigen erst später Symptome, meist während der Nahrungsumstellung auf Breikost. Sie zeigen schwere chronische Verstopfung (Obstipation), Gedeihstörungen, explosionsartige Stuhlentleerungen nach rektaler Untersuchung und in 12-58% der Fälle das Bild eines Darmverschlusses mit Darmentzündung, welche im schlimmsten Fall mit schwerer Erkrankung mit Fieber/ septischem Krankheitsbild und Schocksymptomatik einhergehen kann (toxisches Megakolon).
Wie wird der Morbus Hirschsprung diagnostiziert?
Bei klinischem Verdacht auf einen Morbus Hirschsprung stehen diese diagnostischen Maßnahmen zur Verfügung:
◊ Bei der Röntgenkontrastuntersuchung wird über den Anus Kontrastmittel in den Dickdarm gefüllt und unter Röntgendurchleuchtung untersucht. Ein Darmabschnitt mit Aganglionose stellt sich dabei eng dar mit trichterförmiger Übergangszone in den oft massiv erweiterten „gesunden“ Darm. Die Untersuchung ist relativ strahlenarm durchführbar. Besonders bei Neugeborenen und nach Anlage eines künstlichen Darmausgangs kann es aber zu falsch positiven Befunden kommen.
◊ Bei der anorektalen Manometrie/ Druckmessung kann der Ruhedruck im Rektum gemessen werden. Bei M. Hirschsprung ist dieser meist erhöht (sogenannte Sphinkterachalasie). Die reflektorische Entspannung des inneren Schließmuskels fehlt (fehlende Internusrelaxation) und es zeigen sich keine zum Enddarm gerichtete Darmbewegungen (propulsive Kontraktionen), dafür aber multisegmentale Massenbewegungen. Da die Nervenversorgung beim Neugeborenen noch unreif ist kann die Untersuchung bis zum Alter von 3 Monaten falsch positiv sein. Auch schließt eine normale Relaxation einen Morbus Hirschsprung nicht mit letzter Sicherheit aus.
◊ Die beweisende Untersuchung für die Aganglionose ist die Rektumbiopsie mit anschließender histologischer und immunhistochemischer Untersuchung. Dabei werden mittels einer Enddarmspiegelung Proben der Darmwand in verschiedenen Höhen entnommen. In der feingeweblichen Untersuchung (Histologie) zeigen sich fehlende Ganglienkomplexe (Plexus myentericus und Plexus submucosus) in der Darmwand und vergrößerte cholinerge Nervenfasern. Mittels Immunfärbung werden die Marker Acetylcholinesterase (AChE) und Calretinin dargestellt, welche beim Morbus Hirschsprung vermehrt exprimiert sind.
Wie wird der Morbus Hirschsprung behandelt?
Die Therapie des Morbus Hirschsprung ist immer die Operation. Es gibt eine Vielzahl von Operationsmethoden, wobei allen die vollständige Entfernung des fehlgebildeten Darmsegmentes unter Schonung der Nerven und Muskeln des Beckenbodens gemein ist. Das für Ihr Kind geeignete OP-Verfahren werden wir Ihnen vor der Operation ausführlich erläutern. Die meisten Techniken sind heute auch minimalinvasiv (laparoskopisch gestützt) möglich. Beim akut kranken Kind mit Zeichen eines Darmverschlusses/ toxisches Megakolon kann zunächst die Anlage eines künstlichen Darmausgangs (Anus präter) erforderlich sein. Die endgültige Operation erfolgt dann, wenn das Kind wieder stabilisiert ist. Während der Operation wird mittels Schnellschnittdiagnostik geklärt ob der Absetzungsrand des Darmes sicher Nervenzellen aufweist, also gesund ist.
Welche Komplikationen können auftreten?
In der Frühphase nach der Operation können vor allem Wundinfektionen auftreten. Auch kann es zur Verzögerung der Heilung der Darmneuverbindung (Anastomoseninsuffizienz) kommen. Engstellungen im Bereich der Darmneuverbindung (anorektale Stenosen) können sich entwickeln, was eine Aufweitung erforderlich macht. Durch den Eingriff im Bauchraum können Verwachsungen entstehen, welche einen Darmverschluß verursachen können.
Eine langfristige kinderchirurgische Nachbetreuung in unserer Spezialsprechstunde ist auch zum Erkennen von Spätkomplikationen wichtig. Bis zu 30% der Kinder nach Morbus Hirschsprung leiden unter einer fortbestehenden Verstopfung (Obstipation), Stuhlschmieren sowie Stuhlinkontinenz kommt bei ca. 8% der Patienten vor. Ausserdem können sich Blasenentleerungsstörungen, Engen im Bereich der Darmneuverbindung (Anastomosenstriktur) und fortbestehenden Darmentzündungen entwickeln. Auch wenn sich diese Komplikationen durch keine operative Technik immer verhindern lassen, können wir die Symptome durch ein modernes Darm-Management (Bowel Management - z. B. Einstellung auf Laxantien und Ernährungsberatung bei Verstopfung, die anale Irrigation und Beckenbodentraining bei anhaltender Inkontinenz) lindern und so ein normales soziales Leben ermöglichen.
Urologie
Ureterabgangsstenose
Ureterabgangsstenose
Was ist eine Harnleiterabgangsenge (Ureterabgangsenge)?
Der Urin wird von der Niere ins Nierenbecken abgegeben. Von dort wird er über den Harnleiter zur Harnblase transportiert. Am Abgang des Harnleiters aus dem Nierenbecken kann eine Verengung vorliegen (Ureterabgangsenge). Diese Verengung kann zu einer Aufweitung des Nierenbeckens führen. Eine solche Aufweitung (=Hydronephrose) kann in manchen Fällen bereits vor der Geburt im Ultraschall diagnostiziert werden. Oftmals handelt es sich jedoch um einen Zufallsbefund bei symptomfreien Kindern.
Wie wird eine Ureterabgangsenge diagnostiziert?
Die Aufweitung des Nierenbeckens ist im Ultraschall ausgezeichnet sichtbar. Diese Untersuchung kann ambulant, z.B. im Rahmen unserer kinderurologischen Spezialsprechstunde durchgeführt werden. Je nach Ausprägung des Befundes erfolgt zusätzlich eine Nierenszintigraphie. Diese Untersuchung erlaubt eine seitengetrennte Beurteilung der Nierenfunktion und der Harnabflussverhältnisse.
Um das Zurückfließen des Urins aus der Harnblase in den Harnleiter (vesikoureteraler Reflux) auszuschließen, das ebenfalls mit einer Aufweitung des Nierenbeckens einhergehen kann, ist unter Umständen die Durchführung eines Miktionszysturethrogramms notwendig. Nierenszintigraphie und Miktionszysturethrogramm können ambulant erfolgen.
Wie wird eine Ureterabgangsenge behandelt?
Ist die Funktion der betroffenen Niere durch die Harnstauung nicht eingeschränkt kann eine konservative Behandlung erfolgen. Dazu gehört eine Hohlwegsprophylaxe, also die Einnahme eines niedrig dosierten Antibiotikums zur Verhinderung von Niereninfektionen. Zusätzlich werden regelmäßig Ultraschallkontrollen durchgeführt und der Krankheitverlauf genau dokumentiert.
Bei einer Nierenfunktionseinschränkung aufgrund der Harnstauung erfolgt eine Operation. Es wird eine sogenannte Nierenbeckenplastik durchgeführt. Der Harnleiter wird vom Nierenbecken abgetrennt, die Engstelle entfernt und der gesunde, normal weite Teil des Harnleiters mit dem Nierenbecken verbunden. Diese Operation wird unter Verwendung feinster, resorbierbarer Nahtmaterialien mit der Lupenbrille durchgeführt. Intraoperativ wird eine Schiene, ein sogenannter Doppel-J-Katheter, in den Harnleiter eingebracht um einen problemlosen Urinabfluss zu gewährleisten. Das ist notwendig, da nach der Operation Schwellungen des Gewebes auftreten könne, die den Harnabfluss vom Nierenbecken in die Blase behindern können. Eine Wunddrainage oder Urinableitung nach außen ist nicht notwendig. Für den Patienten und die betreuenden Personen bedeutet das eine erhebliche Erleichterung, da keine externen Drainagen, also nach außen durch die Haut führenden Ableitungen, mehr benötigt werden.
Die Operation kann offen, d.h. durch einen Hautschnitt im Bereich der Flanke, oder minimal invasiv mit der sogenannten Schlüsselloch-Technik durchgeführt werden.
Der Doppel-J-Katheter wird nach ca. 6 Wochen mittels Blasenspiegelung in Kurznarkose entfernt. Die Nierenbeckenplastik ist mit einer Erfolgsrate von ca. 95% eine ausgezeichnete und sichere Methode zur operativen Behandlung der Ureterabgangsenge.
Häufige Fragen:
Muss eine Ureterabgangsenge notfallmäßig operiert werden? - In der Regel handelt es sich NICHT um einen Notfall. Wichtig ist zu erkennen ob eine Einschränkung der Gesamtnierenleistung besteht, ob der Befund einseitig ist und ob Harnwegsinfekte bestehen. Bei Vorhandensein dieser Faktoren kann unter Umständen ein schnelles Vorgehen indiziert sein. Ist die Prophylaxe mit Antibiotika wirklich notwendig und wenn ja, wie lange und schadet diese? - Je nach Befund kann es notwendig sein bei einer Erweiterung der ableitenden Harnwege eine niedrig dosierte Antibiotikaprophylaxe durchzuführen. Durch die niedrige Dosierung treten in der Regel keine ernsthaften Nebenwirkungen und keine vermehrte Infektanfälligkeit bei den Kindern auf.
Wie laufen die Operation und der stationäre Aufenthalt ab? - Die stationäre Aufnahme, auf Wunsch natürlich in Begleitung eines Elternteils, erfolgt am Vortag der OP. Präoperativ werden eine Ultraschalluntersuchung, ein ausführliches Aufklärungsgespräch mit dem Oberarzt und ein Anästhesiegespräch durchgeführt. Die Operation wird in Vollnarkose durchgeführt und dauert ca. 2 Stunden. Postoperativ werden regelmäßige Ultraschallkontrollen durchgeführt. Die Entlassung erfolgt zwischen dem 5. und 7. postoperativen Tag. Der intraoperativ eingelegte Doppel-J-Katheter wird nach 6 Wochen mittels Zystoskopie in Kurznarkose entfernt. Bis dahin wird eine Antibiotikaprophylaxe durchgeführt. Eine Schulbefreiung besteht für ca.1 Woche nach Entlassung und eine Sportbefreiung für 4 Wochen nach Entlassung.
Welche Komplikationen können auftreten und wie erfolgt die Nachkontrolle? - Generell sind Komplikationen sehr selten. Zu nennen sind Infektion und Wundheilungsstörung, aber auch spezielle Komplikationen wie das Austreten von Urin im Bereich der Naht und erneute Verengung des Ureters durch Narbenbildung. Die engmaschige Nachkontrolle erfolgt in unserer Spezialsprechstunde zur Beurteilung des Nierenwachstums und Erkennen von ev. Folgeerkrankungen.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Uretermündungsstenose
Uretermündungsstenose
Was ist ein Megaureter?
Der Urin wird von der Niere ins Nierenbecken abgegeben. Von dort wird er über den Harnleiter zur Harnblase transportiert. Man spricht von einer Erweiterung des Harnleiters, wenn dessen Durchmesser ≥ 6 mm beträgt. Ursachen für eine Harnleitererweiterung sind (1) eine Engstelle am Übergang des Harnleiters in die Harnblase, (2) das Zurückfließen des Urins aus der Harnblase in den Harnleiter (vesikoureterorenaler Reflux) oder (3) eine Gewebsschwäche, die dazu führt, das sich der Harnleiter nicht zusammenziehen kann und so mit Urin aufgefüllt und gedehnt wird. Dabei kann es zu einem Rückstau von Urin bis ins Nierenbecken ( = Hydronephrose) kommen.
Eine Harnleitererweiterung mit oder ohne Erweiterung des Nierenbeckens kann in manchen Fällen bereits vor der Geburt im Ultraschall diagnostiziert werden. Oftmals handelt es sich jedoch um einen Zufallsbefund bei symptomfreien Kindern.
Wie wird ein Megaureter diagnostiziert?
Die Aufweitung des Harnleiters mit oder ohne Erweiterung des Nierenbeckens ist im Ultraschall sichtbar. Diese Untersuchung kann ambulant, z.B. im Rahmen unserer kinderurologischen Spezialsprechstunde durchgeführt werden. Je nach Ausprägung des Befundes erfolgt zusätzlich eine Nierenszintigraphie. Diese Untersuchung erlaubt eine seitengetrennte Beurteilung der Nierenfunktion und der Harnabflussverhältnisse. Um das Zurückfließen des Urins aus der Harnblase in den Harnleiter (vesikoureteraler Reflux) auszuschließen, das ebenfalls mit einer Aufweitung des Harnleiters einhergehen kann, ist unter Umständen die Durchführung eines Miktionszysturethrogramms notwendig. Nierenszintigraphie und Miktionszysturethrogramm können ambulant erfolgen.
Wie wird ein Megaureter behandelt?
Ist die Funktion der betroffenen Niere durch die Harnstauung nicht eingeschränkt kann eine konservative Therapie erfolgen. Dazu gehört eine Hohlwegsprophylaxe, also die Einnahme eines niedrig dosierten Antibiotikums zur Verhinderung von Niereninfektionen. Zusätzlich werden regelmäßig Ultraschallkontrollen durchgeführt und der Krankheitsverlauf genau dokumentiert.
Bei einer Nierenfunktionseinschränkung aufgrund der Harnstauung erfolgt eine Operation. Zunächst wird der Harnleiter von der Blase abgetrennt und im Bereich der Bauchdecke ausgeleitet. Der Urin kann einfach abfließen und Harnleiter sowie Niere können sich erholen. Der Patient wird nach Abschluss der Wundheilung nach Hause entlassen. Es erfolgen engmaschige Ultraschallkontrollen. Zusätzlich wird über einen Katheter der Druck im ausgeleiteten Harnleiter gemessen. Diese Untersuchungen werden ambulant durchgeführt. Erreicht der Druck im Harnleiter wieder normale Werte erfolgt eine Rückverlagerung des Harnleiters, also ein erneuter Anschluss an die Harnblase. Diese Operation findet ca. 12 Monate nach dem ersten Eingriff statt. Der Anschluss des Harnleiters an die Harnblase (Uretrocystoneostomie) wird unter Verwendung feinster, resorbierbarer Nahtmaterialien mit der Lupenbrille durchgeführt. Intraoperativ wird eine Schiene, ein sogenannter Doppel-J-Katheter, in den Harnleiter eingebracht um einen problemlosen Urinabfluss zu gewährleisten. Das ist notwenig, da nach der Operation Schwellungen des Gewebes auftreten können, die den Harnabfluss vom Harnleiter in die Blase behindern können.
Der Doppel-J-Katheter wird nach ca. 6 Wochen mittels Blasenspiegelung in Kurznarkose entfernt. Die Uretrocystoneostomie ist mit einer Erfolgsrate von ca. 90 % eine ausgezeichnete und sichere Methode zur Wiederherstellung der Kontinuität im Bereich des oberen Harntraktes.
Häufige Fragen:
Muss ein Megaureter notfallmäßig operiert werden? - In der Regel handelt es sich NICHT um einen Notfall. Wichtig ist zu erkennen ob eine Einschränkung der Gesamtnierenleistung besteht, ob der Befund einseitig ist und ob Harnwegsinfekte bestehen. Bei Vorhandensein dieser Faktoren kann unter Umständen ein schnelles Vorgehen indiziert sein.
Ist die Prophylaxe mit Antibiotika wirklich notwendig und wenn ja, wie lange und schadet diese? - Je nach Befund kann es notwendig sein bei einer Erweiterung der ableitenden Harnwege eine niedrig dosierte Antibiotikaprophylaxe durchzuführen. Durch die niedrige Dosierung treten in der Regel keine ernsthaften Nebenwirkungen und keine vermehrte Infektanfälligkeit bei den Kindern auf.
Wie laufen die Operation und der stationäre Aufenthalt ab? - Die stationäre Aufnahme, auf Wunsch natürlich in Begleitung eines Elternteils, erfolgt am Vortag der OP. Präoperativ werden eine Ultraschalluntersuchung, ein ausführliches Aufklärungsgespräch mit dem Oberarzt und ein Anästhesiegespräch durchgeführt. Die Operation wird in Vollnarkose durchgeführt und dauert ca. 2 Stunden. Postoperativ werden regelmäßige Ultraschallkontrollen durchgeführt. Die Entlassung erfolgt zwischen dem 5. und 7. postoperativen Tag. Der intraoperativ eingelegte Doppel-J-Katheter wird nach 6 Wochen mittels Zystoskopie in Kurznarkose entfernt. Bis dahin wird eine Antibiotikaprophylaxe durchgeführt.
Welche Komplikationen können auftreten und wie erfolgt die Nachkontrolle? - Generell sind Komplikationen sehr selten. Zu nennen sind Infektionen und Wundheilungsstörung, aber auch spezielle Komplikationen wie das Austreten von Urin im Bereich der Naht und erneute Verengung des Ureters durch Narbenbildung. Die engmaschige Nachkontrolle erfolgt in unserer Spezialsprechstunde zur Beurteilung des Nierenwachstums und Erkennen von ev. Folgeerkrankungen. Die Aufweitung des Harnleiters mit oder ohne Erweiterung des Nierenbeckens ist im Ultraschall sichtbar. Diese Untersuchung kann ambulant, z.B. im Rahmen unserer kinderurologischen Spezialsprechstunde durchgeführt werden. Je nach Ausprägung des Befundes erfolgt zusätzlich eine Nierenszintigraphie. Diese Untersuchung erlaubt eine seitengetrennte Beurteilung der Nierenfunktion und der Harnabflussverhältnisse. Um das Zurückfließen des Urins aus der Harnblase in den Harnleiter (vesikoureteraler Reflux) auszuschließen, das ebenfalls mit einer Aufweitung des Harnleiters einhergehen kann, ist unter Umständen die Durchführung eines Miktionszysturethrogramms notwendig. Nierenszintigraphie und Miktionszysturethrogramm können ambulant erfolgen.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Vesikoureteraler Reflux
Vesikoureteraler Reflux
(=Rückfluss von Harn aus der Blase über den Harnleiter in das Nierenbecken)
Was ist ein VUR?
Der Urin wird von der Niere ins Nierenbecken abgegeben. Von dort wird er über den Harnleiter zur Harnblase transportiert. Der Übergang Harnleiter-Harnblase ähnelt einem Ventil, welches zulässt, dass Urin in die Blase gelangt, dieser Urin aber nicht zurück in den Harnleiter fließen kann. Beim Wasserlassen steigt der Druck in der Harnblase. Bei einer Funktionseinschränkung am Übergang Harnleiter-Harnblase kann es dazu kommen, dass Urin zurück in den Harnleiter fließt. Bei einer schweren Funktionsstörung kann der Urin bis ins Nierenbecken zurückfließen und zu einer Erweiterung des Harnleiters und Nierenbeckens (= Hydronephrose) führen. Klinisch fallen die Kinder oft durch mehrfach aufgetretene Harnwegsinfekte als Folge des Refluxes auf. Harnweginfekte oder im schlimmsten Fall eine Niereninfektion sind unter Umständen schwierig zu diagnostizieren. Bei unklarem Fieber und Trinkschwäche, Bauchschmerzen und/ oder Schmerzen beim Wasserlassen sollte auf jeden Fall eine Urinuntersuchung mit Frage nach einer Infektion stattfinden.
Wie wird ein VUR diagnostiziert?
Um das Zurückfließen des Urins aus der Harnblase in den Harnleiter darzustellen, ist die Durchführung eines Miktionszysturethrogramms (MCU) notwendig. Dabei wird über einen Blasenkatheter die Harnblase mit Kontrastmittel gefüllt und unter Röntgenkontrolle der Urinabfluss dokumentiert. Besteht am Übergang Harnleiter-Harnblase eine Funktionsstörung, ein vesikoureteraler Reflux, fließt auch das Kontrastmittel zurück in den Harnleiter, was mit Hilfe einer Röntgenaufnahme sicher nachgewiesen werden kann. Das Miktionszysturethrogramm wird ambulant durchgeführt. Eine Aufweitung des Harnleiters mit oder ohne Erweiterung des Nierenbeckens ist im Ultraschall sichtbar. Diese Untersuchung wird ebenfalls ambulant, z.B. im Rahmen unserer kinderurologischen Spezialsprechstunde, durchgeführt. Je nach Ausprägung des Befundes erfolgt zusätzlich eine Nierenszintigraphie. Diese Untersuchung erlaubt eine seitengetrennte Beurteilung der Nierenfunktion, die Aufgrund des Refluxes beeinträchtigt sein kann.
Wie wird ein VUR behandelt?
Bei auftreten von Harnwegsinfektionen und nachgewiesenem Reflux erfolgt zunächst eine konservative Therapie. Dazu gehört eine Hohlwegsprophylaxe, also die Einnahme eines niedrig dosierten Antibiotikums zur Verhinderung von Niereninfektionen. Zusätzlich werden regelmäßig Ultraschallkontrollen durchgeführt und der Krankheitsverlauf genau dokumentiert. Oftmals bildet sich, vor allem während der ersten Lebensjahre, der Reflux spontan zurück. Bei einer Nierenfunktionseinschränkung oder bei wiederkehrenden Infektionen unter Hohlwegsprophylaxe erfolgt eine Operation. Dabei können, je nach Schweregrad des Refluxes, verschiedene Techniken zur Anwendung kommen. Ein leichtgradiger Harnreflux, der unter Einnahme eines niedrig dosierten Antibiotikums immer wieder zu Infektionen führt, kann minimal invasiv, d.h. durch eine Blasenspiegelung und Unterspritzen der Ureteröffnung zur Verengung derselben, erfolgen. Bei hochgradigem, schwerem Reflux ist oftmals eine offene Operation die einzige Therapiemöglichkeit. Dabei wird der Harnleiter zunächst von der Blase abgetrennt und unter Bildung eines suffizienten, neuen Ventilmechanismus erneut in die Blase eingebaut. Der Anschluss des Harnleiters an die Harnblase (Uretrocystoneostomie) wird unter Verwendung feinster, resorbierbarer Nahtmaterialien mit der Lupenbrille durchgeführt. Intraoperativ wird eine Schiene, ein sogenannter Doppel-J-Katheter, in den Harnleiter eingebracht um einen problemlosen Urinabfluss zu gewährleisten. Das ist notwenig, da nach der Operation Schwellungen des Gewebes auftreten können, die den Harnabfluss vom Harnleiter in die Blase behindern können. Der Doppel-J-Katheter wird nach ca. 6 Wochen mittels Blasenspiegelung in Kurznarkose entfernt. Die Uretrocystoneostomie ist mit einer Erfolgsrate von ca. 90 % eine ausgezeichnete und sichere Methode zur Therapie des hochgradigen Harnrefluxes.
Häufige Fragen:
Muss ein VUR notfallmäßig operiert werden? - In der Regel handelt es sich NICHT um einen Notfall. Wichtig ist bei nachgewiesenen, wiederkehrenden Niereninfektionen rasch mit einer Hohlwegsprophylaxe, also der Einnahme eines niedrig dosierten Antibiotikums zur Verhinderung von neuen Infektionen zu beginnen. Sollte eine chirurgische Therapie notwendig sein, kann diese geplant erfolgen.
Ist die Prophylaxe mit Antibiotika wirklich notwendig und wenn ja, wie lange und schadet diese? - Bei nachgewiesenen, wiederkehrenden Niereninfektionen aufgrund eines Harnrefluxes ist eine niedrig dosierte Antibiotikaprophylaxe durchzuführen. Niereninfektionen führen zu Narbenbildungen im Bereich des Nierengewebes. Das wiederum kann eine Funktionseinschränkung bis hin zum Funktionsverlust der Niere zur Folge haben. Weiterhin ist das Auftreten von Bluthochdruck nach mehrfachen Niereninfektionen eine gefürchtet Komplikation.
Durch die niedrige Dosierung des Antibiotikum treten in der Regel keine ernsthaften Nebenwirkungen und keine vermehrte Infektanfälligkeit bei den Kindern auf.
Wie laufen die Operation und der stationäre Aufenthalt ab? - Die stationäre Aufnahme, auf Wunsch natürlich in Begleitung eines Elternteils, erfolgt am Vortag der OP. Präoperativ werden eine Ultraschalluntersuchung, ein ausführliches Aufklärungsgespräch mit dem Oberarzt und ein Anästhesiegespräch durchgeführt. Die Operation wird in Vollnarkose durchgeführt und dauert je nach Eingriff 1 bis 2 Stunden. Postoperativ werden regelmäßige Ultraschallkontrollen durchgeführt. Die Entlassung erfolgt zwischen dem 5. und 7. postoperativen Tag. Der intraoperativ eingelegte Doppel-J-Katheter wird nach 6 Wochen mittels Zystoskopie in Kurznarkose entfernt.
Welche Komplikationen können auftreten und wie erfolgt die Nachkontrolle? - Generell sind Komplikationen sehr selten. Zu nennen sind Infektion und Wundheilungsstörung, aber auch spezielle Komplikationen wie eine Verengung des Ureters durch Narbenbildung oder ein Rezidiv, d,h, ein postoperativ weiter besehender Harnrückfluss in den Ureter. Die engmaschige Nachkontrolle erfolgt in unserer Spezialsprechstunde zur Beurteilung des Nierenwachstums und Erkennen von ev. Folgeerkrankungen. Nach ca. 6 Monaten erfolgt ambulant ein Kontrollmiktionszyst-urethrogramm.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Hypospadie
Hypospadie
(=Fehlmündung der Harnröhre)
Was ist eine Hypospadie?
Eine Hypospadie ist eine angeborene Fehlbildung der Harnröhre. Diese endet nicht an der Spitze der Glans sondern an der Unterseite des Penis. Oft besteht dadurch eine schlechte Kontrollierbarkeit des Harnstrahls. Zusätzlich kann eine Penisschaftkrümmung (Chordee) auftreten, die je nach Ausmaß bei Erektion Schmerzen verursachen und zu einer deutlichen Einschränkung beim Geschlechtsverkehr führen kann. Die psychosoziale Entwicklung des Kindes, insbesondere in der Pubertät, kann dadurch deutlich negativ beeinflusst werden.
Wie wird eine Hypospadie diagnostiziert?
Bei der Hypospadie handelt es sich um die häufigste urogenitale Fehlbildung beim Jungen. Etwa 5 von 1000 männlichen Säuglingen sind davon betroffen. Die Diagnose erfolgt nach der Geburt. Es zeigt sich eine sogenannte dorsale Präputialschürze, d.h. die Vorhaut im Bereich der Vorderseite des Penis ist wie eine Schürze vergrößert und bedeckt die Glans. Beim genauen Hinsehen entdeckt man die Fehlmündung der Harnröhre an der Unterseite des Penis. Meistens handelt es sich um eine kleine, punktförmige Öffnung. Beim Wasserlassen fließt der Harnstrahl aus dieser Fehlmündung.
Wie wird eine Hypospadie behandelt?
Der ideale OP-Zeitpunkt liegt zwischen dem 12. und 18. Lebensmonat. Die meisten Hypospadien (ca. 85%) treten im Bereich des distalen Penisschaftes auf. Bei der Operation wird der fehlende Teil der Harnröhre plastisch rekonstruiert und die Penisschaftverkrümmung korrigiert. Der Blasenkatheter zur Schienung der neuen Harnröhre wird bereits nach 48 Stunden entfernt. Eine Harnableitung durch die Bauchdecke ist nicht notwendig. In unserer Klinik kann sich der Patient nach der OP in vollem Umfang bewegen und muss nicht mehr im Bett fixiert werden. Der Krankenhausaufenthalt beträgt nur vier Tage.
Häufige Fragen:
Muss eine Hypospadie notfallmäßig operiert werden? - Es handelt sich NICHT um einen Notfall. Wichtig ist eine Sonographie der Nieren und ableitenden Harnwege um andere Fehlbildungen auszuschließen. <
Wie laufen die Operation und der stationäre Aufenthalt ab? - Die stationäre Aufnahme, auf Wunsch natürlich in Begleitung eines Elternteils, erfolgt am Vortag der OP. Präoperativ werden eine Ultraschalluntersuchung, ein ausführliches Aufklärungsgespräch mit dem Oberarzt und ein Anästhesiegespräch durchgeführt. Die Operation wird in Vollnarkose durchgeführt und dauert ca. 2 Stunden. Die Entlassung erfolgt am 3. postoperativen Tag.
Welche Komplikationen können auftreten und wie erfolgt die Nachkontrolle? - Die häufigste postoperative Komplikation ist eine Harnfistel, d.h. der Urin fließt nicht aus der neu konstruierten Harnröhrenöffnung an der Spitze der Glans sondern durch eine Öffnung im Bereich der Naht. Weltweit beträgt diese Komplikationsrate 5% - 20%. Als weitere mögliche Komplikation ist die Verengung der Harnröhre zu nennen. Die engmaschige Nachsorge mit regelmäßiger Harnstrahldruckmessung und Restharnbestimmung erfolgt in unserer kinderurologischen Sprechstunde.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Harnwegsinfekt
Harnwegsinfekt
Was ist ein Harnwegsinfekt?
Bei Harnwegsinfektionen handelt es sich um die häufigsten bakteriellen Entzündungen im Kindesalter. Ca. 1% der Jungen und 3-5% der Mädchen haben in ihrer Kindheit Harnwegsinfektionen. Diese könne sich nur auf die unteren Harnwege also die Blase beschränken (Zystitis) oder aufsteigend die oberen Harnwege also das Nierenbecken und die Niere selbst betreffen (Pyelonephritis). Klinisch äußert sich die Blasenentzündung (Zystitis) durch Schmerzen bei Wasserlassen (Dysurie), gehäuftes Wasserlassen (Pollakisurie), Harndrang und Schmerzen im Unterbauch. Fieber kann ebenfalls vorkommen. Bei der Nierenbeckenentzündung (Pyelonephritis) kommt es neben Flankenschmerzen zu einem Klopfschmerz der Flanke und meist hohem Fieber. Bei Neugeborenen und Säuglingen können auch Symptome wie Nahrungsverweigerung, Gereiztheit oder übermäßige Schläfrigkeit, „unklare“ Fieberepisoden oder Fieberkrämpfe, Erbrechen und Durchfall Hinweis für eine Harnwegsinfektion sein.
Wie wird ein Harnwegsinfekt diagnostiziert?
Die Harnwegsinfektion wird mittels einer Urinuntersuchung auf weiße Blutkörperchen (Leukozyten), rote Blutkörperchen (Erythrozyten), Eiweiß (Protein) und Bakterien gesichert. Beweisend ist dabei das Wachstum von Bakterien in einer Urinkultur. Zur Urinentnahme ist bei älteren „trockenen“ Kindern ein Mittelstrahlurin ausreichend, bei kleineren Kindern kann eine Einmalkatheterisierung erforderlich sein. Bei Verdacht auf einen oberen Harnwegsinfekt (Pyelonephritis) ist die Untersuchung der Entzündungswerte im Blut sinnvoll.
Insbesondere bei der ersten Harnwegsinfektion ist zusätzlich eine Ultraschalluntersuchung der Nieren und ableitenden Harnwege erforderlich, da bei bestimmten angeborene Formen der Harntransportstörung (Vesikoureteraler Reflux, Uretermündungsstenose (Megaureter) und seltener auch die Ureterabgangsstenose) ein erhöhtes Risiko für Harnwegsinfektionen besteht. Da aufsteigende Harnwegsinfektionen (Pyelonephritiden) zu einer narbigen Schädigung der Nieren und damit zu deren Funktionsminderung oder -Verlust führen können ist es wichtig diese behandelbaren Ursachen zu erkennen. Bei kleinen Säuglingen oder nach dem zweiten fieberhaften Harnwegsinfekt sollte daher auch eine Miktionszysturethrographie (MCU) zum Ausschluß eines Vesikoureteralen Refluxes erfolgen. Bei Verdacht auf Harnabflußstörungen ist eine Nierenszinitigraphie zur Berurteilung des Harnabflusses und der Nierenfunktion notwendig.
Wie wird ein Harnwegsinfekt behandelt?
Zur Behandlung der Harnwegsinfektionen stehen verschiedene Antibiotika zur Verfügung, die richtige Auswahl trifft ihr behandelnder Arzt. Bei unteren Harnwegsinfektionen (Zystitiden) können die Antibiotika als Saft oder Tabletten (oral) eingenommen werden. Bei oberen Harnwegsinfektionen (Pyelonephritiden) ist in der Regel eine intravenöse antibiotische Kombinationstherapie über 7-14 Tage erforderlich.
Insbesondere bei Harntransportstörungen wie dem höhergradigen vesikoureteralen Reflux und der Uretermündungsstenose (Megaureter) sollte in den ersten 6-12 Lebensmonaten und nach einer Harnwegsinfektion ein geeignetes niedrig dosiertes Antibiotikum als Harnwegsinfektprophylaxe eingenommen werden.
Auch wenn eine konservative Therapie in vielen Fällen möglich ist, kann in Einzelfällen (z.B. gehäuft auftetenden Harnwegsinfektionen trotz antibiotischer Prophylaxe, bereits bestehender Schädigung einer Niere, etc.) ein operativer Eingriff zur Behebung der Harntransportstörung erforderlich werden. (Siehe Vesikoureteraler Reflux, Megaureter, Ureterabgangsstenose) . Bei bereits trockenen Kindern ist auch ein Toilettentraining zur Behebung von Blasenentleerungstörungen als Prophylaxe wichtig. Bei Vorhautverengungen kann die Vorhaut als Keimreservoir für Harnwegsinfektionen dienen, so dass diese ebenfalls durch sparsame Beschneidung behoben werden sollten.
Wir in der Universitätsmedizin Greifswald arbeiten in einem interdisziplinären Team aus Kinderurologen und Kindernephrologen um die für ihr Kind jeweils optimale konservative wie operative Therapie zu gewährleisten.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
neurogene Blase
neurogene Blase
Was ist eine neurogene Blase?
Die neurogene Blase des Kindes ist überwiegend eine Folge angeborener Neuralrohrdefekte in Folge einer Schlussstörung im Wirbelsäulenbereich. In seltenen Fällen kann eine neurogene Blasenfunktionsstörung im Kindesalter auch die Folge von Traumata, Entzündungen und Tumoren sein. Hierbei kommt es zu einer Fehlfunktion der Blasenmuskulatur und der Schließmuskulatur. Es kann sich als unfreiwilliger Urinverlust (Harninkontinenz) oder als Schwierigkeit die Blase zu entleeren (Blasenentleerungsstörung) darstellen.
Die Blase dient als Speicher für den von den Nieren produzierten Urin. Eine Funktionsstörung der Blase kann auf Dauer - und im schlimmsten Fall zu einer nicht mehr behandelbaren Schädigung der Nieren führen.
Therapieziele:
Die Therapieziele einer neurogenen Blasenfunktionsstörung sind:
1. Schutz bzw. Verbesserung der Nierenfunktion
2. Optimierung der Blasenentleerung
3. Harnkontinenz
Um eine optimale Therapie gewährleisten zu können ist die Art der Fehlfunktion von Blase und Schließmuskel sowie die Funktion von Nieren und Harnleiter mittels diagnostischer Untersuchungen zu ermitteln. Hierzu gehören die Ultraschalluntersuchung, Urinuntersuchung, Blasendruckmessung, Miktionscystourethrogramm und Szintigramm. Je nach Art der Fehlfunktion stehen konservative und operative Therapieformen zur Verfügung. Konservative Behandlungsformen wären z.B. Einmalkatheterismus, Pharmakotherapie und Blaseninstillation. Operative Therapien umfassen z.B. die Einspritzung von Botulinum Toxin (Botox) in die Muskulatur der Blase, Blasenaugmentation und die kontinente kutane Harnableitung.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Traumatologie
Verbrennungen
Verbrennungen
Definition:
Verbrühung als häufigste Ursache mit Altersgipfel von 1-3 Jahren. Thermische Schädigung der Haut unterschiedlichen Schwere- und Tiefengrades.
Grade:
Wir unterscheiden Grad I mit Hautrötung bis Grad IV Verkohlung. Dazwischen liegen Grad II und III mit unterschiedlich tiefer Schädigung. Ab Grad III ist mit einer Narbenbildung zu rechnen.
Notfallmaßnahmen:
Sofortige Kühlung unter kaltem Wasser (20°C für 20 Minuten), um ein Tieferbrennen zu verhindern. Nachfolgend steriler Verband (keine Salben usw.). Schmerztherapie mit Schmerzzäpfchen zB. Paracetamol-Supp. Nachfolgend Vorstellung in Kinderchirurgischer Ambulanz (Impfausweis nicht vergessen). Grossflächige Verletzungen sollten über den Notarzt zur Klinik gebracht werden.
Therapie:
I. gradige Verletzungen bedürfen in aller Regel eines Salbenverbandes auf desinfizierender Basis (z.B.: Octenigel®). Je nach Ausmass der Verletzung ist eine stationäre Überwachung notwendig. Empfehlenswert ist diese bei Verletzungen von über 5% Körperoberfläche, des Gesichtes oder des Genitale. Aufgrund großer Flüssigkeitsverluste muß bei größeren Flächen eine Infusionsbehandlung erfolgen.
Verbrennungsblasen sollten abgetragen und ggf. gedeckt werden. Dazu eignen sich Amnion als auch Suprathel® zum zeitweiligen Hautersatz. Tiefer gehende Verletzung müssen chirurgisch mittels Nekrektomie (Entfernung abgestorbener Hautanteile) und Hauttransplantation versorgt werden. Nachfolgend empfiehlt sich eine gute Kompressions- und / oder Silkonsalbenbehandlung zur Vermeidung unschöner hypertropher Narbenbildung. Gerade bei Verletzungen der Hände, Füße bzw. gelenkübergreifend an großen Gelenken ist unter Umständen später eine Narbenkorrektur zur Vermeidung oder Behebung von Fehlstellungen nötig.
Häufige Fragen:
Wie lange muß behandelt werden? - In Abhängigkeit von Größe und Tiefe der Verletzung kann sich eine Behandlung von ca. 7 bis 10 Tagen oder länger hinziehen.
Hat mein Kind Schmerzen? - Verbrennungen oder Verbrühungen sind sehr schmerzhaft. Durch eine bedarfsgerechte Schmerzmittelgabe versuchen wir diese so niedrig wie möglich zu halten.
Kann ich mein Kind mit betreuen? - Unbedingt, denn das trägt zur schnelleren Genesung bei.
Bleiben entstellende Narben zurück? - Die tiefergradigenVerletzungen sind durch Defektheilung und Narbenbildung charakterisiert. Durch kompetentes Management kann jedoch das Ausmaß eingeschränkt werden. Dazu gehören die frühzeitige Nachbehandlung mittels Kompression und Silikonsalbe.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Klavikulafrakturen
Klavikulafrakturen
Die Clavikulafraktur ist mit ca. 1,7 % die häufigste geburtstraumatische Schädigung des Neugeborenen. Im Kindesalter umfasst sie ca. 5-15% der Frakturen und ist damit die vierthäufigste Frakturform in dieser Altersgruppe. Entsprechend anderer traumatischer Verletzungen, sind Jungen häufiger als Mädchen betroffen. Als klassischer Unfallmechanismus wird zumeist der direkte Sturz auf die Schulter beschrieben. Geburtstraumatischen Clavikulafrakturen werden zumeist durch den sich bildenden Kallus mit einer konsekutiven Schwellung klinisch apparent. Wesentlich seltener fallen sie durch einen asymmetrischen Moro-Reflex oder ein asymmetrisches Stillverhalten auf. Im Kindesalter führen zumeist die typische Anamnese für das Unfallgeschehen, eine typische Klinik inkl. der typischen Schonhaltung der oberen Extremität sowie in vielen Fällen eine sicht- bzw. palpable Deformierung der Clavikula zur Verdachtsdiagnose. Aufgrund des im Kindesalter noch kräftig ausgebildeten Periostschlauchs kommt es in dieser Altersgruppe selten zu Dislokationen, so dass eine Fehlstellung der Clavikula häufig fehlt. In der Adoleszenz hingegen überwiegen die dislozierten Clavikulafrakturen. Als gebräuchlichste Klassifikation der Clavikulafrakturen im Kindesalter gilt die nach Allmann, welche die Frakturen hinsichtlich ihrer Lokalisation einteilt. Unterteilt wird diesbezüglich in die Frakturen des mittleren Clavikuladrittels (Typ I; >90%), Frakturen des lateralen Clavikuladrittels (Typ II; 5%) und die des medialen Claviculadrittels (Typ III; ca. 3%). Das Diagnostikum der Wahl ist zwar die Röntgenuntersuchung, allerdings können Frakturen des mittleren Drittels auch ausschließlich sonographisch diagnostiziert werden. In Einzelfällen ist bei Verdacht auf eine mediastinale Dislokation im Rahmen einer Fraktur des medialen Clavikuladrittels ein MRT zum Ausschluss einer konsekutiven Kompression mediastinaler Strukturen indiziert. Clavikulafrakturen haben aufgrund ihres hohen Korrekturpotentials eine sehr gute Prognose und können in der Mehrzahl der Fälle konservativ mittels Ruhigstellung behandelt werden (Gilchrist- oder Rucksackverband). Absolute Operationsindikation stellen offene Frakturen, eine bereits bestehende Perforation bzw. die Perforationsgefahr, Verletzungen neurovaskulärer Strukturen sowie eine Kompression mediastinaler Strukturen dar. Relative Operationsindikationen wären beispielsweise eine erhebliche Verkürzung der Clavikula, ein möglicherweise störendes kosmetisches Ergebnis und eine hohe sportliche Aktivität des Kindes. Insgesamt ist die operative Versorgung zumeist eine Abwägung zwischen Vor-und Nachteilen bzw. Nutzen und Risiken der Eingriffe, welche mit den Eltern unter Erläuterung der etwaigen Risiken besprochen werden muss. Optionen der operativen Versorgung sind in Abhängigkeit der Lokalisation eine ESIN-Osteosynthese (Nagel) oder die Zuggurtungsosteosynthese. Das Therapieziel, sowohl der konservativen als auch der operativen Versorgung, ist die komplette Wiederherstellung der Schulterbeweglichkeit unter Verwendung möglichst einfacher Methoden und kurzer Rekonvaleszenzzeiten.
Plastisch-rekonstruktive Chirurgie
Hämangiom
Hämangiom
Definition:
- Gefäßfehlbildung infolge Fehlsteuerung während der Embryogenese
- Häufigkeit von ca. 3% aller Säuglinge autretend
Symptome:
- bunt vielfältiges Erscheinungsbild (Blutgefäßknäuel an der Hautoberfläche oder auch unter der Haut oder in Körperhöhlen und inneren Organen liegend)
- unterschiedlichste Formen von "Pünktchen" bis flächiger Ausbreitung
- an unterschiedlichesten Körperteilen liegend
- nach Geburt auftretend mit Größenwachstum mit und ohne Blutung
- spontane Rückbildung möglich, aber auch rasche Größenzunahme mit starker Beeinträchtigung
- Formen: Feuermahl, Blutschwämmchen, Hämangiom, Spidernaevi, Portwein Stain u.a.
Diagnostik:
- "Blickdiagnose" beim Spezialisten
- Größenvermessung
- Fotodokumentation
- ggf. Ultraschalluntersuchung
- Verlaufsbeobachtung durch Anbindung an Spezialsprechstunde
- bei "inneren" Hämangiomen apparative Diagnostik wie Angio-MRT, Blasen- oder Darmspiegelung
- ggf. Gerinnungsdiagnostik bei multiplen Auftreten oder sehr großen Formen
(Kasabach Merritt Syndrom)
Behandlungsbedarf / konservative Behandlung:
- nicht alle Hämangiome sind behandlungspflichtig, da eine Rückbildungstendenz im
1. oder bis zum 6. Lebensjahr besteht - bei ausgeprägter Größenprogredienz mit Befall wichtiger Strukturen, Entstellung oder Irritationsneigung ist eine Behandlung notwendig
Operation:
Früher wurden viele Hämangiome alleine durch eine Operation therapiert. Dies hatte eine mitunter entstellende Narbe zur Folge. Heute wird ein multimodales Therapiekonzept gefahren.
Nach Diagnostik werden die meisten Hämangiome primär im Verlau beobachtet. Kritische Hämangiome können einer Lasertherapie (Farbstofflaser und Neodym-YAG-Laser) zugeführt werden. Damit können diese ohne große Narbenbildung geheilt werden. Sehr tief gehende Hämangiome können auch punktiert und nachfolgend von innen (interstitiell) gelasert werden. Diese Behandlung muß wiederholt werden.
Extrem schnell wachsende und entstellende Hämangiome an Gesicht, Körperstamm und Extremitäten können uU. mit einem Blutdruck senkendem Medikament (Propranolol) behandelt werden. Dies stellt derzeit noch keine Leitlinien konforme Behandlungsstrategie dar, kann aber im Einzelfall nach vorausgegangener kardiologischer Diagnostik nach Absprache mit den Eltern vorgenommen werden.
Führen große Gefäße in das Hämangiom ist mitunter eine Embolisation notwendig. Dabei werden die versorgenden Gefäße unter röntgenologischer Kontrolle verödet. Nachfolgend kann die Lasertherapie fortgesetzt und oder eine plastisch chirurgische Therapie angewandt werden.
Ergebnisse:
- überwiegend gute Ansprechrate der Therapie
- bei Lasertherapie mehrere Sitzungen im Intervall notwendig
- wenig Komplikationen durch Eiswürfeltechnik unter Schonung der Haut
Häufige Fragen:
Wie oft muss behandelt werden? - In Abhängigkeit von der Größe ca. 3-4 mal.
Hat mein Kind Schmerzen? - Durch die Laserung in Kurznarkose, Vorkühlung beim Lasern und Gabe vom Schmerzmitteln kaum.
Kann ich mein Kind betreuen? - Ja, wir haben täglich großzügiges Besuchsrecht oder auch die Möglichkeit der stationären Mitaufnahme der Mütter.
Ist die Narkose für mein Kind gefährlich? - Wir haben erfahrene Kinderanästhesisten, welche täglich Kinder betreuen.
Wer darf Lasern? - Ärzte mit Zertifikat nach Besuch eines Laserkurses und erworbenen klinischen Erfahrungen.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Lymphangiome
Lymphangiome
Definition:
Von den Lymphbahnen ausgehende Fehlbildung unter Ausbildung von klein- bis großzystischen Hohlräumen durch ausbleibende oder fehlerhafte Verbindung mit einem zentralen lymphatischen oder venösen Sammelsystem.
Symptome / Formen:
- Zystische Raumforderung an Kopf und der Hals (zusammen ca. 50 %), Axilla (13%),
- Rumpf (8 %) und Extremitäten mit 11%. Seltener sind Lymphangiome im Abdomen, Retroperitoneum, Mediastinum, Knochen oder Skrotum zu finden.
- Lymphangioma simplex: Kleine Zysten aus erweiterten lymphatischen Kapillaren nicht nur an der Haut, sondern auch an der Zunge, am Gaumen, den Schleimhäuten, am Genitale und an den Skleren, in Operationsnarben, meist oberflächlich liegend (Warzen oder in Gruppen von Bläschen), oft mit Blutungen oder Krusten versehen, mitunter zur Selbstheilung neigend nach Blaseneruption.
- Lymphangioma cavernosum: Im lockeren Unterhautfettgewebe liegende, häufig auch subserös im Mesenterium, aber auch im Retroperitoneum und im Mediastinum liegend. Es besteht aus solitären, gelegentlich septierten meist aber aus multiplen Zysten von unterschiedlicher Grösse mit lymphatischem Endothel.
- Lymphangioma cysticum: Es sind vorwiegend kleinzystische, solide erscheinende Geschwülste, die infiltrierend wachsen und in die Organe einbrechen. Sie sind dadurch schwer abgrenzbar, bestehend aus vielen dilatierten Lymphgefässen, welche eine ein- oder mehrlagige Epithelauskleidung der Wände zeigen. Haemolymphangiom, ein Mischtumor, der zusätzlich hämangiomatöse (von den Blutgefäßen ausgehende) Anteile aufweist.
Diagnostik:
- Ultraschall: entweder als multizystisch, septierte Masse ohne Fluss außer in den Septen (grosszystische Ly.) oder als hyperechogene Struktur ohne intralesionärem Fluss im Doppler (mikrozystische Ly.).
- MRT: zeigt eine tiefe Signalintensität in der T1- Wichtung und eine hohe Signalintensität in der T2 Wichtung, Varianz bei Einblutung mit differenter Intensität
Angiographischen Verfahren wie die Lymphographie, Venographie und Angiographie bringen keine zusätzlichen Informationen.
Behandlungsbedarf:
Einblutung, Entzündung, Ruptur oder Exulzeration sind häufige Komplikationen. Beeinträchtigungen durch die Raumforderung mit Organschädigungen wie Schluck- und Atemstörungen, Fehlstellungen der Wirbelsäule, des Knochens oder Kiefers, Frakturen sowie Chylaskos, Chyloperikard und Chylothorax können infolge auftreten.
Therapie:
Da zum Zeitpunkt der Diagnose bereits eine oder mehrere Komplikationen bestehen und die Lymphangiome einerseits keine spontane Regression zeigen und andererseits die Kinder häufig aufgrund der Komplikation gefährdet sind, ist eine möglichst rasche und radikale Behandlung anzustreben, um den Komplikationen vorzubeugen. Dies gilt auch für die asymptomatischen Formen
- 1. radikale Operation:
- 2. medikamentöse systemische Therapie:
- Kortikosteroide
- Interferon α 2A
- Zytostatika
- 3. Lokaltherapie:
- Sklerosierungstherapie
- Lokale Vaccination (Bleomycin , Äthoxysklerol, Ethibloc, Fibrin, Alkohol,
Essigsäure, Glucose 50%, NaCl 20% , OK432 (Streptococcus pyogenes Gruppe A
Typ III 3 mit Benzylpenicillin behandelt)
- 4. Lasertherapie:
- Koagulation direkt perkutan (durch die Haut)
- Koagulation intraläsionär (LITT)
- Vaporisation (Ablation)
- Resektion, Fensterung
Ergebnisse:
- überwiegend gutes Ansprechen bei rechtzeitiger Therapie
- mitunter Rezidive (Rückfälle)
- nicht immer radikal behandelbar, da sonst benachbarte Strukturen beeinträchtigt werden
Häufige Fragen:
Wie oft muß behandelt werden? - In Abhängigkeit von der Methode (Sklerosierung, Vaccination, Laser) mehrfach. Operativ kann ein Eingriff ausreichend sein. Nicht jedes Lymphangiom bietet sich jedoch für eine solche Behandlung an. Der Arzt sollte nach Diagnostik aus dem ganzen Spektrum die für ihr Kind optimale mitunter auch Kombinationstherapie auswählen.
Hat mein Kind Schmerzen? - Unser Motto ist ein schmerzfreies Krankenhaus, deshalb nehmen wir ein Schmerzmonitoring vor und verordnen individuell bedarfsgerecht Schmerzmittel
Kann ich mein Kind betreuen? - Unbedingt, denn das trägt zur schnelleren Gesundung bei. Wir haben ein großzügiges Besuchsrecht oder die Möglichkeit der Mitaufnahme der Mütter oder Väter.
Ist die Narkose für mein Kind gefährlich? - Wir haben erfahrene Kinderanästhesisten welche im täglichen Umgang mit Kindern geübt sind.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
konservative Behandlungen
chronische Bauchschmerzen
chronische Bauchschmerzen
Alle Kinder haben irgendwann mal Bauchschmerzen, diese sind im Kindesalter die häufigste Schmerzursache. Akut auftretende Bauchschmerzen sollten immer rasch abgeklärt werden.
Definition:
Chronische, rezidivierende Bauchschmerzen haben ein breites Feld an Diagnosen, die in Frage kommen könnten. Die Schmerzen können organischer oder funktioneller Natur sein. In den meisten Fällen handelt es sich dabei um funktionelle Bauchschmerzen. Eine organische Ursache sollte vor Diagnosestellung trotzdem immer ausgeschlossen werden. Die Anamnese und die klinische Untersuchung sind bei chronischen Bauchschmerzen ein sehr wichtiger Bestandteil des Arztbesuches. Durch die Fragen und Untersuchungsbefunde kann der Arzt meist schon eine Reihe von Diagnosen ausschließen.
Symptome:
Das Hauptsymptom chronischer Bauchschmerzen sind rezidivierende Bauchschmerzen mit monatlich mehreren auftretenden Episoden über mindestens 3 Monate, die mit einer Störung der normalen täglichen Aktivitäten des Kindes einhergehen. Häufig treten zusätzlich bei organischen Ursachen für die Bauchschmerzen folgende Symptome auf:
- Veränderung der Stuhlgewohnheiten / Stulkonsistenz
- Blut / Schleim im Stuhl
- Überlkeit
- Erbrechen
- Kopfschmerzen
- Fieber
- Ausschlag / Hautveränderungen
- Gelenkschmerzen / Gelenkschwellung
- rezidivierende Aphten im Mund
- Leistungsknick
- raschere Ermüdbarkeit
- Gewichtsverlust
- Wachstumsverzögerungen
Diagnostik:
Die nach der klinischen Untersuchung eventuell durchzuführende Diagnostik hängt von der Verdachtsdiagnose des Arztes ab und reicht von Ultraschall über Blutentnahme, Urinuntersuchung, Stuhluntersuchung, Atemtests, radiologische Untersuchung, bis hin zu
Magen-, Darm- und explorativer Bauchspiegelung. Es muss jedoch zwischen dem Nutzen der Untersuchung und der Belastung, die dadurch für das Kind entsteht, abgewogen werden.
Behandlung:
Die meisten organischen Erkrankungen können konservativ mit Diät oder Medikamenten behandelt werden. Es gibt jedoch einige Erkrankungen die eine langfristige Therapie bedürfen. In den seltenen Fällen ist eine Operation zur Behebung der Grunderkrankung notwendig. Bei funktionellen Beschwerden sollte ein möglichst normaler Alltag gestaltet werden. Dabei erfolgt die Behandlung konservativ (ggf. Wärmflasche / Tee...). Die Kinder müssen eine Strategie zum Umgang mit den Schmerzen entwickeln. In schwierigen Fällen kann die Betreuung durch einen Kinderpsychologen hilfreich sein.
Häufige Fragen:
Was kann ich als Elternteil tun, um den Arztbesuch so effektiv wie möglich zu gestalten? - Sie können mit Ihrem Kind zusammen vor dem Arztbesuch einige Wochen ein Beschwerdeprotokoll führen und dieses zum Arztbesuch mitbringen. Die kann schon erste Hinweise für den Arzt liefern
Gibt es Anzeichen, die eher auf eine organische Ursache hinweisen? - Ja, die gibt es. Darunter fallen die obengenannten Begleitsymptome. Des weiteren gehören dazu Bauchschmerzen, die zu nächtlichen erwachen des Kindes führen; eine Schmerzlokalisation vom Nabel entfernt, anhaltende Schmerzen über Minuten/Stunden, so wie ausstrahlende Schmerzen und familiäre Belastung mit intestinalen Erkrankungen dazu.
Kontakt:
Kinderchirurgie
Telefon: 03834 - 86 7037
Fax: 03834 - 86 7038
Email:kinderchirurgie@med.uni-greifswald.de
Obstipation
Obstipation
Definition:
Eine Obstipation (Verstopfung) kann am besten als Stuhlretention (Stuhlverhaltung) infolge unvollständiger Stuhlentleerung beschrieben werden. Alle Kinder haben irgendwann mal Stuhlgangsprobleme im Sinne einer Obstipation. Eine wichtige Rolle spielt dabei wie lange diese andauert. Von einer chronischen Obstipation spricht man bei einer Beschwerdedauer von mehr als 2 Monaten. Eine Obstipation kann mehrere Ursachen haben. Man unterscheidet zwischen selten auftretenden organischen und häufig auftretenden funktionellen Ursachen.
Deshalb ist die Anamnese und die klinische Untersuchung, vor allem des Rektums und der Afterregion, bei Obstipation ein sehr wichtiger Bestandteil des Arztbesuches. Durch die Fragen und Untersuchungsbefunde kann der Arzt meist schon eine Reihe von Diagnosen ausschließen.
Symptome:
Die Hauptsymptome, die sich aus einer Stuhlretention ergeben sind vielfältig. Darunter fallen Bauchschmerzen, meist rezidivierend und kurz anhaltend. Des weiteren zählen dazu Defäkationsschmerz, oft großkalibriger und harter Stuhl, unwillkürlicher Stuhlabgang, Analfissuren, perinale Entzündungen, Blutauflagerungen auf dem Stuhl, Übelkeit, Erbrechen, Blähungen und ausladendes Abdomen.
Diagnostik:
Die nach der klinischen Untersuchung eventuell durchzufühende Diagnostik hängt von der Verdachtsdiagnose des Arztes ab und reicht von der Urinuntersuchung über Blutentnahme, anorektale Manometrie, Kolon-Kontrasteinlauf, Rektoskopie/Sigmoidskopie mit Probenentnahme bis hin zu Ultraschall, radiologischer Untersuchung, Defäkographie, MRT und Kolontransitzeit. Es muss jedoch zwischen dem Nutzen der Untersuchung und der Belastung, die dadurch für das Kind entstehen, abgewogen werden.
Behandlung:
Die meisten organischen Erkrankungen können konservativ behandelt werden. In den seltesten Fällen ist eine Operation zur Behebung der Grunderkrankung notwendig. Bei funktioneller Ursache kann teilweise schon mit einer ballaststoffreichen Ernährung und einer Trinkmenge von mindestens 1,5-2 liter pro Tag und Toilettentraining eine Verbesserung erreicht werden. Bei schwerer und schon länger andauernden Obstipation ist fast immer eine initiale Darmentleerung durch z.B. Einläufe oder Zäpfchen notwendig. Daran sollte sich immer eine Erhaltungstherapie meist mit oralen Medikamenten anschließen. Diese muss oft Wochen bis Monate lang durchgeführt werden, dass der Darm sich erholen kann.
In schwierigen Fällen kann die Betreuung durch einen Kinderpsychologen hilfreich sein. Das Ziel der Behandlung ist eine schmerzfreie regelmäßige Defäkation zu erreichen.
Häufige Fragen:
Was kann ich als Elternteil tun um den Arztbesuch so effektiv wie möglich zu gestalten? - Sie können mit Ihrem Kind zusammen vor dem Arztbesuch für einige Wochen ein Stuhlprotokoll führen und dieses zum Arztbesuch mitbringen. Dies kann dann schon erste Hinweise für den Arzt liefern.
Wodurch wird die funktionelle Obstipation verursacht? - Manchmal findet sich primär keine identifizierbare Ursache. Bei Kindern können jedoch folgende Ursachen vorliegen:
- unausgewogene Ernährung
- geringe Flüssigkeitszufuhr
- Bewegungsmangel
- Ignorieren des Stuhldranges und "vergessen" auf die Toilette zu gehen
- unterdrücken des Stuhldranges bei schmerzhafter Stuhlentleerung
- psychisch-emotionale Faktoren
Warum ist es so wichtig, eine Obstipation zu behandeln? - Bei beginnender Obstipation kann sich leicht eine Art "Teufelskreis" entwickeln: harter Stuhl führt zu schmerzhafter Stuhlentleerung, wodurch wiederum ein Stuhlverhalten durch das Kind resultiert. Durch den Stuhlverhalt kann es so zur Ansammlung großer und harter Stuhlmengen im Rektum kommen, was einen verminderten Dehungsreiz und damit eine verminderte Wahrnehmungsschwelle des Stuhldranges zur Folge hat.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Tumorchirurgie
Neuroblastom
Neuroblastom
Das Neuroblastom ist die häufigste bösartige Tumorerkrankung des Kindesalters. Der Tumor geht vom Nervensystem aus und verläuft in den meisten Fällen von der Nebenniere oder vom Nervengeflecht neben der Wirbelsäule in den Bauch- und Brustraum. Zwei Drittel der Kinder sind bei Auftreten der Erkrankung jünger als ein Jahr.
Das Neuroblastom bildet Absiedlungen v.a. Lymphknoten, Knochenmark, Leber und Haut.
Symptome:
Das Neuroblastom ist häufig ein Zufallsbefund, der bei Ultraschalluntersuchung entdeckt wird. Oft fällt auch den Eltern oder dem Kinderarzt ein großer tastbarer Tumor im Bauch oder eine Verwölbung der Bauchdecke auf. Der Tumor verursacht zunächst Allgemeinsymptome wie Schmerzen, Durchfall, Appetitlosigkeit und ausbleibende Gewichtszunahme. Je nach Lokalisation des Tumors kann es auch zu anderen Symptomen wie Husten oder Auftreten einer Nierenstauung kommen.
Diagnostik:
Besteht der Verdacht auf ein Neuroblastom bei einem Kind, werden zunächst das Blut und das Knochenmark untersucht. Außerdem erfolgt eine genaue Bildgebung mit Ultraschall- und Kernspinuntersuchung. Durch die Kinderchirurgie wird eine Biopsie vorgenommen. Dabei werden mindestens zwei Gewebeproben entnommen, die dann genauestens untersucht werden.
Behandlung:
Therapiemöglichkeiten beim Neuroblastom sind die operative Tumorentfernung, Chemotherapie und Bestrahlung. Meist nutzt man eine Kombination dieser Verfahren, abhängig vom Tumorstadium und der Risikogruppe des jeweiligen Kindes. Aufgaben der Kinderchirurgie sind hierbei neben der Probenentnahme die Entfernung des Tumors durch Operation, aber auch die Entfernung von Lymphknoten und ggf. die chirurgische Behandlung.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Nephroblastom
Nephroblastom
Das Nephroblatom ist der häufigste Nierentumor im Kindesalter. Etwa 100 Kinder erkranken pro Jahr in Deutschland neu an dieser bösartigen Erkrankung. Bei 5-10 % der betroffenen Kinder geht der Tumor von beiden Nieren aus. Der Erkrankungsgipfel liegt im 2. und 3. Lebensjahr.
Symptome:
Das Nephorblastom bleibt meist lange völlig symptomlos und fällt schließlich durch eine Schwellung im Flankenbereich sowie durch eine tastbare Vorwölbung des Tumors auf. Selten treten Bauchschmerzen, Fieber, Schwäche oder Blut im Urin auf.
Diagnostik:
Das Nephroblastom ist gut durch Ultraschall darstellbar. Außerdem werden laborchemische Untersuchungen von Blut und Urin vorgenommen. Die Kernspintomographie dient der Darstellung des Tumors, mit einem Röntgenbild der Lunge wird nach möglichen Tumorabsiedlungen gesucht. Weitere Untersuchungen können ausgeschlossen werden. Bei Verdacht auf ein Nephroblastom erfolgt keine Probenentnahme aus dem Tumorgewebe, da hierbei die Gefahr bestünde, Tumorzellen zu verschleppen. Durch die moderne Bildgebung kann ein Nephroblastom mit ausreichender Sicherheit diagnostiziert werden.
Behandlung:
In Europa erfolgt bei Kindern mit Nephroblastom zunächst eine präoperative Chemotherapie. Dadurch soll der Tumor kleiner werden, außerdem sinkt die Gefahr, dass es zu einer Tumorruptur kommt. Nach Abschluss dieser Chemotherapie erfolgt als Erfolgskontrolle und für die Planung der Operation eine erneute Bildgebung. Bei der Operation wird dann der Tumor radikal entfernt. Es wird außerdem die betroffene Niere i.A. komplett entfernt. Sind beide Nieren vom Tumor befallen, wird versucht, so viel wie möglich gesundes Nierengewebe zu belassen. Außerdem werden bei der Operation natürlich die Lymphknoten im Bauchraum untersucht. Im Anschluss an die Operation kann in Abhängigkeit vom Tumorstadium eine weitere Chemotherapie und eine Strahlentherapie stattfinden.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Hepatoblastom
Hepatoblastom
In Mitteleuropa erkranken 2 von 1 Million Kindern an einem Hepatoblastom, dem häufigsten Lebertumor im Kindesalter. Der Altersgipfel liegt zwischen einem halben Jahr und drei Jahren. Tumorabsiedlungen außerhalb der Leber treten meist erst spät im Erkrankungsverlauf auf.
Symptome:
Symptome eines Hepatoblastoms können ein tastbarer Tumor im rechten Oberbauch, Fieber, Bauchschmerzen, Gewichtsverlust und Veränderungen des Blutbildes sein.
Diagnostik:
Die Bildgebung bei Verdacht auf ein Hepatoblastom besteht im Ultraschall, Computer- und Kernspintomographie der Leber und einer Röntgenaufnahme der Lunge. Im Blut können bestimmte Hormone nachgewiesen werden, die bei einem Hepatoblastom erhöht sind. Ist die Diagnose nicht sicher, werden zusätzlich bei einer Operation Gewebeproben entnommen.
Behandlung:
Die operative Entfernung des Hepatoblastoms erfolgt im Anschluss an eine Chemotherapie. Auch nach der Operation muss die Chemotherapie fortgeführt werden. Eine Starhlentherapie wird beim Hepatoblastom i.A. nicht angwendet. Bei sehr ausgedehnten Tumoren kann auch eine Lebertransplantation nötig sein.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
Rhabdomyosarkom
Rhabdomyosarkom
Das Rhabdomyosarkom ist der häufigste Weichteiltumor im Kindesalter und entsteht aus einem Vorläufergewebe der Muskulatur. Am häufigsten tritt dieser Tumor im Bereich von Kopf und Hals auf, gefolgt von Tumoren der Harnwege oder Geschlechtsorgane. Er kommt aber auch am Rumpf und an den Extremitäten vor. Es gibt zwei Häufigkeitsgipfel mit einem Auftreten bis zum 7. Lebensjahr oder im Jugendalter.
Symptome:
Das Erscheinugnsbild der Erkrankung ist abhängig vom Sitz des Tumors. Im Allgemeinen kommt es zu einer Schwellung sowie zu einer Beeinträchtigung der benachbarten Strukturen. So kann ein Tumor im Kopfbereich zum Beispiel zu Nasenbluten, Gesichtslähmung oder Sehstörungen führen.
Diagnostik:
Im Anschluss an eine genaue Bildgebung erfolgt die Tumorbiopsie durch die Kinderchirurgie. Außerdem ist eine Knochenmarkuntersuchung notwendig.
Behandlung:
Der Zeitpunkt der Operation beim Rhabdomyosarkom ist abhängig von der Lokalisation des Tumors und seiner Ausdehnung. In manchen Fällen kann der Tumor direkt nach Diagnosestellung komplett entfernt werden, oft findet die Entfernung des Tumors aber auch erst nach einer Chemotherapie statt. Auch die Strahlentherapie kommt bei Weichteiltumoren zum Einsatz.
Kontakt:
Kinderchirurgie
Telefon: 03834-86 7037
Fax: 03834-86 7038
Email: kinderchirurgie@med.uni-greifswald.de
ambulante Operationen
Hydrozele
Hydrozele
Definition:
Bei einer Hyrocele oder Wasserbruch wird die Ansammlung von Flüssigkeit um den Hoden (im Hodensack) bezeichnet. Wenn sich eine Wasseransammlung im Hodensack befindet, spricht man von einer Hydocele testis. Wenn sich diese im Inguinalbereich befindet, dann spricht man von einer Hydrocele funikuli.
Klinische Symptomatik
Prall-elastische Schwellung des Hodensacks ohne Infektzeichen (Rötung und Überwärmung).
Diagnostik:
- Diaphonoskpie
Es wird der Hodensack durchgeleuchtet - Sonografie
Therapie:
- Abwarten bis zum 1. Lebensjahr, weil ein Spontanverschluß möglich ist
- Operation
- wenn das Kind >1 Jahr ist und kein Spontanverschluß vorliegt
- wenn das Kind eine sehr große Hydrocele (v.a. bei Säuglingen) oder eine kommunizierende Hydrocele mit wechselnder Größe hat
- wenn das Kind einen begleitenden Leistenbruch hat.